Table of Contents
- 1 Viruses Required for Complex Life:
- 2 Viral Sequences make up Majority of Human Genome:
- 3 Where did our Genomic Viral Sequences Come From?
- 4 The Argument for Common Descent:
- 5 Potential Inconsistencies with Common Descent Arguments:
- 5.1 Most Viral Sequences are Non-Functional:
- 5.2 Most ERVs are Species Specific:
- 5.3 Most Retroviruses Insert Randomly within a Genome:
- 5.4 A Nested Hierarchical Pattern:
- 5.5 Shared Mistakes are Random:
- 5.6 Genomic Fixation:
- 5.7 Origin of ERVs and other Retroviruses:
- 6 Summary:
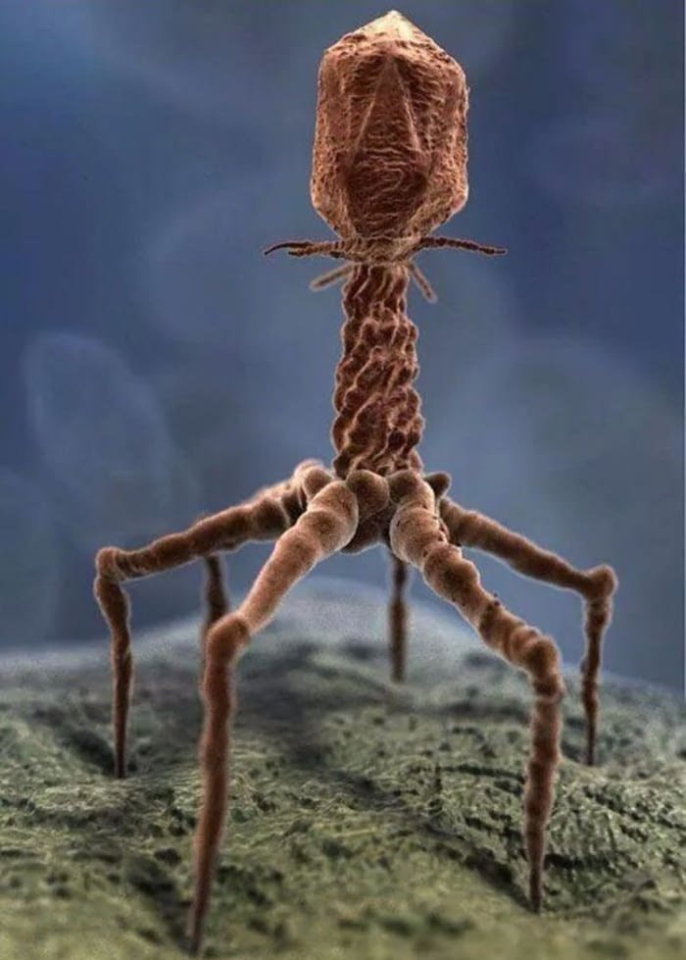 Viruses Required for Complex Life:
Viruses Required for Complex Life:
The knee-jerk reaction when hearing the word “virus” is an almost universal negative reaction for most people. After all, viruses are associated with a host of benign as well as severe sicknesses and diseases from the common cold to HIV and its associated autoimmune deficiency syndrome or AIDS.
Yet, upon closer examination, it turns out that most viruses are good – even vital for the existence of complex life such as human life. They help to clean contaminated rivers (like the Ganges River in India which has, for centuries, been revered for its “self-cleansing and special healing properties”). They are vital for intestinal health in humans and animals (Link). The average person absorbs up to 30 billion phages every day through the intestines (Link). In fact, most of the human body isn’t actually human. Human cells make up only 43% of the body’s total cell count (Link). The rest are microscopic colonists, comprised by at least 38 trillion bacteria and over 380 trillion viruses – collectively known as the “human virome” (Link). And, of these colonists, most have never been studied and are currently unknown to science. Up to 52% of the DNA fragments isolated from a given human fail to match anything in existing genetic databases (Link). The chronic inflammation associated with such diseases as inflammatory bowel diseases (i.e., Crohn’s disease and ulcerative colitis), cancer, Alzheimer’s, autism, diabetes, various heart conditions, rheumatoid arthritis and more, is associated with the lack of a well-balanced virome. This means that the use of bacteriophages that are known to solely target harmful bacteria that plague mammals and humans, open new ways to prevent and cure these diseases (Link, Link, Link).
Viral Sequences make up Majority of Human Genome:
 What is particularly interesting, in this light, is that viruses are, apparently, even more intimately associated with humanity. Their genetic sequences actually make up the majority of the human genome – 52.1% based on the most recent reference genome sequences (Tang, 2019). There are different types of these viral-type sequences, or mobile genetic elements. For instance, about 8% of the human genome is composed of endogenous retroviruses (ERVs). Short Interspersed Nuclear Elements (or SINEs) contribute ∼13% of the human genome and Long Interspersed Nuclear Elements (or LINEs) contribution ∼17% to the human genome by sequence length… etc.
What is particularly interesting, in this light, is that viruses are, apparently, even more intimately associated with humanity. Their genetic sequences actually make up the majority of the human genome – 52.1% based on the most recent reference genome sequences (Tang, 2019). There are different types of these viral-type sequences, or mobile genetic elements. For instance, about 8% of the human genome is composed of endogenous retroviruses (ERVs). Short Interspersed Nuclear Elements (or SINEs) contribute ∼13% of the human genome and Long Interspersed Nuclear Elements (or LINEs) contribution ∼17% to the human genome by sequence length… etc.
Where did our Genomic Viral Sequences Come From?
 So, where did these sequences come from? Scientists generally propose that these viral-type genetic sequences gradually infiltrated the ancestral genome(s) of all modern humans, apes, and mammals and other plants and animals in general, over the course of many millions of years of time (Link). In fact, is is concluded that viruses have been infecting vertebrates and inserting themselves in vertebrate genomes “for over 450 million years” (Link). The ages of these genetic sequences can be determined using mutational distances from each phylogenetic tree branch to find the rate of molecular evolution at each particular locus for a genetic viral sequence.
So, where did these sequences come from? Scientists generally propose that these viral-type genetic sequences gradually infiltrated the ancestral genome(s) of all modern humans, apes, and mammals and other plants and animals in general, over the course of many millions of years of time (Link). In fact, is is concluded that viruses have been infecting vertebrates and inserting themselves in vertebrate genomes “for over 450 million years” (Link). The ages of these genetic sequences can be determined using mutational distances from each phylogenetic tree branch to find the rate of molecular evolution at each particular locus for a genetic viral sequence.
This conclusion seems rather obvious. After all, viruses are built to invade and take over other cells as a means for their own reproduction – right? It only makes sense, then, that the viral elements present within the human genome would have arrived there via the same mechanism of innumerable ancestral viral infections and genomic integrations over the course of the vast periods of evolutionary time on this planet. In fact, this process seems so obvious that many evolutionary biologists, and evolutionists in general, believe that the existence of ERVs and other viral genetic elements within the genomes of living things, forming a nested hierarchical tree-like pattern between species, is one of the best evidences of the reality of common descent. Consider, for example, the following passage from the current Wiki and RationalWiki articles on ERVs.
However, where viruses first came from or how they came to be is not apparent from an evolutionary perspective. It would seem self-evident that these viruses would have had to originally arise within an independent free-living life form – since all viruses depend on living cells in order to replicate. A virus cannot replicate independent from a living cell, which supplies it with energy and mechanisms for replication.
The Argument for Common Descent:
Because most HERVs have no function, are selectively neutral, and are very abundant in primate genomes, they easily serve as phylogenetic markers for linkage analysis (Wiki, 2019).
ERVs are usually species-specific, inserted almost randomly in the host genome, and the error or mutation that inactivated the gene is random. If two organisms share the same ERV in the same location with the same inactivation mutations, then they almost certainly share them due to common inheritance and not two separate infections. Researchers analyze shared ERV insertions across species to construct phylogenetic trees. For example, the common ERVs in simians indicates they share a common genome. When phylogenetic trees are constructed based on the pattern of ERVs, they indicate humans share more ERVs with chimps than either share with gorillas. Other examples are known. This is strong evidence for common descent. (RationalWiki, 2019)
Domestication of the syncytin genes represents a dramatic example of convergent evolution via the cooption of a retroviral gene for a key biological function in reproductive biology. In fact, syncytin domestication from a retroviral envelope gene has been previously shown to have independently occurred at least seven times during mammalian evolution (Malik, 2012).
In other words, for those who would think to argue for intelligent design for the origin and diversity of life on this planet, why would any intelligent designer insert viral genetic sequences into humans, apes, and other plants and animals? – sequences that are largely non-functional and broken in similar and even identical ways in closely related species (such as humans and apes)? Such a proposal simply makes no sense. At the very least the evidence in hand seems to neatly fit the theory of common descent – so much so as to be a very solid nail in the coffin for intelligent design advocates. Yet, things are not always as they might first appear…
Potential Inconsistencies with Common Descent Arguments:
Most Viral Sequences are Non-Functional:
Because most HERVs have no function, are selectively neutral, and are very abundant in primate genomes, they easily serve as phylogenetic markers for linkage analysis (Wiki, 2019).
 More and More all the Time:
More and More all the Time:
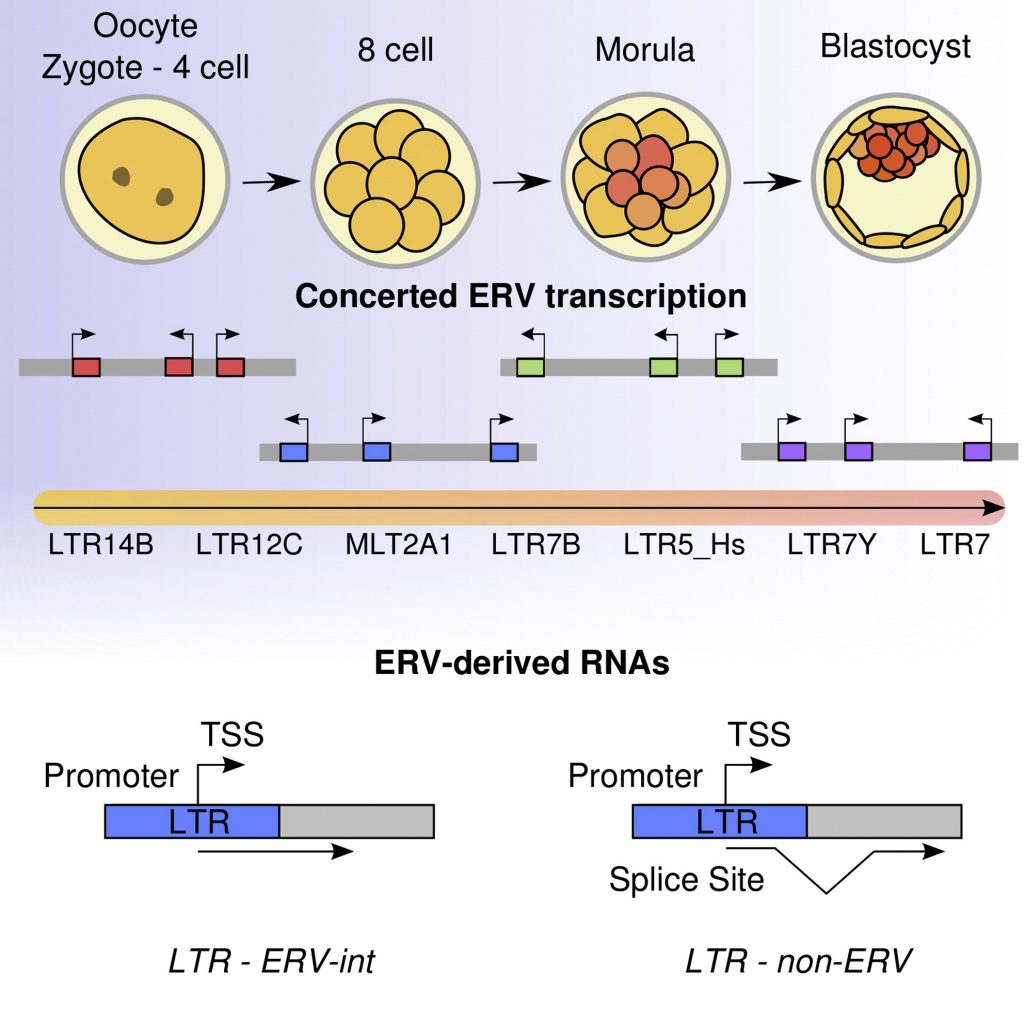
The problem here, as it turns out, is that more and more discoveries are suggesting that a significant percentage, and probably most ERVs and other mobile genetic viral-type elements within the human genome, are actually functional to one degree or another. Many are even vital for the performance of a host of different key functional features in different species. It isn’t just a few here or there that have somehow evolved some functionality over vast periods of time. Rather, such sequences seem to form the very basis for the unique form and function of humanity and other species. The protein-coding genes themselves are not really where the key information is stored within the genome. Rather, these basic “genes” function more like simple building blocks, bricks and mortar if you please, that can be used to build a wide variety of structures with very different functions. Where the real higher-level blueprint information is contained is within the non-coding regions of the DNA – largely within viral-type sequences like ERVs and other mobile elements.
In a 2013 study, Ward et al. demonstrated that under a heterologous (derived from a different species) regulatory environment, regulatory sites in mobile elements, including those specific to humans, can be activated to alter histone modifications and DNA methylation (i.e., epigenetic control) as well as expression of nearby genes in both germline and somatic cells (Ward, 2013).
We report the existence of 51,197 ERV-derived promoter sequences that initiate transcription within the
human genome, including 1,743 cases where transcription is initiated from ERV sequences that are located in gene proximal promoter or 5’ untranslated regions (UTRs)…. Our analysis revealed that retroviral sequences in the human genome encode tens-of-thousands of active promoters; transcribed ERV sequences correspond to 1.16% of the human genome sequence, and PET tags that capture transcripts initiated from ERVs cover 22.4% of the genome” (Conley, et al., 2008, 24[14]:1563,1566).A profound implication of this observation is that lineage- and species-specific mobile or transposable elements (MEs or TEs) can provide novel regulatory sites to the host genome, which can potentially regulate nearby genes’ expression in a lineage- and species-specific manner and lead to phenotypic differences. A very recent study added such an example by showing that an ERV element is responsible for regulating innate immunity in humans by controlling the expression of adjacent IFN-induced genes…
A total of 7,547 or 50.7% of these HS-MEs [Human-specific MEs) are located inside or in the 1 kb promoter regions of genes for protein coding, non-coding RNAs, as well as transcribed pseudogenes (Table 3), which represent 4,607 unique genes/transcripts (data not shown)…
Outside of the promoter and exon regions, 1,167 of the HS-MEs contribute to 3,032 binding sites for 142 of the 161 examined transcriptional factors. While their specific functional impact would be hard to predict computationally and can only be more accurately assessed/validated experimentally, many examples of such functional impact have been demonstrated (Tang, 2019).
ENCODE Findings:
As far as the findings of the ENCODE project are concerned:
Using DNase I hypersensitivity data sets from ENCODE in normal, embryonic, and cancer cells, we found that 44% of open chromatin regions were in TEs and that this proportion reached 63% for primate-specific regions. We also showed that distinct subfamilies of endogenous retroviruses (ERVs) contributed significantly more accessible regions than expected by chance, with up to 80% of their instances in open chromatin. Based on these results, we further characterized 2,150 TE subfamily-transcription factor pairs that were bound in vivo or enriched for specific binding motifs, and observed that TEs contributing to open chromatin had higher levels of sequence conservation. We also showed that thousands of ERV-derived sequences were activated in a cell type-specific manner, especially in embryonic and cancer cells, and we demonstrated that this activity was associated with cell type-specific expression of neighboring genes. Taken together, these results demonstrate that TEs, and in particular ERVs, have contributed hundreds of thousands of novel regulatory elements to the primate lineage and reshaped the human transcriptional landscape.
For example, we observed that 1237 of the 2337 (52.9%) LTR7 repeat instances (a subfamily of the LTR/ERV class) were contributing to open chromatin in the human embryonic stem cell (ESC) line H7 when we would have only expected 60.5 (2.6%). This corresponds to a 20-fold enrichment and is highly significant (p<1.0E-100). We call such repeat subfamilies DHS-associated repeats (DARs) … although LTR/ERV repeats constitute 13.5% of the repeat instances in the genome, they represent 25.0%, 54.6%, and 33.0% of the DAR instances in normal, embryonic, and cancer cells, respectively. … LTR/ERV repeats have contributed a disproportionate fraction of cell type-specific accessible chromatin regions especially in embryonic and cancer cell lines. This is interesting given that network rewiring using ERV elements has already been described in ESCs [embryonic stem cells] and that it has been shown that stem cell potency fluctuates with endogenous retrovirus activity in mouse. … Finally, we also reported that repeat subfamilies activated in a cell type-specific manner were also frequently associated with higher expression of neighboring genes.
(Pierre-Étienne Jacques, Justin Jeyakani, Guillaume Bourque, “The Majority of Primate-Specific Regulatory Sequences Are Derived from Transposable Elements,” PLOS Genetics (May 9, 2013)
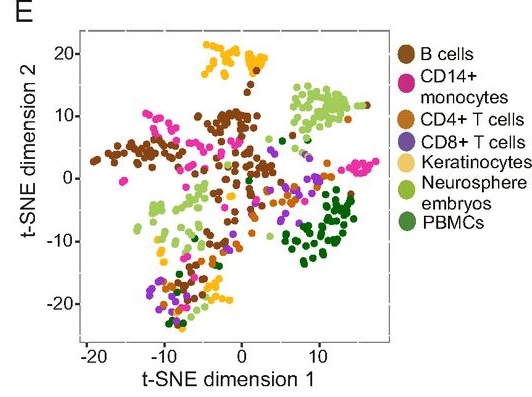 Other more recent studies have shown the same thing – that specific cell types can be identified based on the types of ERVs that are activated within that specific cell type. Amazingly, “ERV expression is unique enough to allow discrimination between cell types. Similar to cell lines, roughly 50% of the ERV loci were expressed by any given cell type. All cells expressed similar total levels of ERV transcripts, but distinct sets of ERVs were transcribed in a given cell type.” (Maria Tokuyama et. al., 2018)
Other more recent studies have shown the same thing – that specific cell types can be identified based on the types of ERVs that are activated within that specific cell type. Amazingly, “ERV expression is unique enough to allow discrimination between cell types. Similar to cell lines, roughly 50% of the ERV loci were expressed by any given cell type. All cells expressed similar total levels of ERV transcripts, but distinct sets of ERVs were transcribed in a given cell type.” (Maria Tokuyama et. al., 2018)
In other words, ERVs are being transcribed in a highly non-random manner that correlates with specific types of cell functionality as well as embryological patterns in association with other functional genetic elements. This seems to clearly point towards important functionality for many and probably most ERVs. Sure, some of this functionality may be redundant functionality, but redundancy is often part of designed systems that show enhanced resistance to breakdown – such as in computer programming. In any case, functionally redundant sequences do not negate their functional characteristics (Pitman, 2014 and Pitman, 2018)
More Opinion than Fact:
We would submit that differential expression (including extensive alternative splicing) of RNAs is a far more accurate guide to the functional content of the human genome than logically circular assessments of sequence conservation, or lack thereof. Assertions that the observed transcription represents random noise (tacitly or explicitly justified by reference to stochastic (‘noisy’) firing of known, legitimate promoters in bacteria and yeast), is more opinion than fact and difficult to reconcile with the exquisite precision of differential cell- and tissue-specific transcription in human cells (for a recent debate see van Bakel et al. 2010; Clark et al. 2011). Moreover, where tested, these noncoding RNAs usually show evidence of biological function in different developmental and disease contexts, with, by our estimate, hundreds of validated cases already published and many more en route, which is a big enough subset to draw broader conclusions about the likely functionality of the rest. It is also consistent with the specific and dynamic epigenetic modifications across most of the genome, and concurs with the ENCODE conclusion that 80% of the genome shows biochemical indices of function (Dunham et al. 2012). Of course, if this is true, the long-standing protein-centric zeitgeist of gene structure and regulation in human development will have to be reassessed (Mattick 2004, 2007, 2011), which may be tacitly motivating the resistance in some quarters. (Mattick and Dinger, 2013)
But, what about the “C-value Enigma”?
The other substantive argument that bears on the issue, alluded to in the quotes that preface the Graur et al. article, and more explicitly discussed by Doolittle (Doolittle 2013), is the so-called ‘C-value enigma’, which refers to the fact that some organisms (like some amoebae, onions, some arthropods, and amphibians) have much more DNA per cell than humans, but cannot possibly be more developmentally or cognitively complex, implying that eukaryotic genomes can and do carry varying amounts of unnecessary baggage. That may be so, but the extent of such baggage in humans is unknown. However, where data is available, these upward exceptions appear to be due to polyploidy and/or varying transposon loads (of uncertain biological relevance), rather than an absolute increase in genetic complexity (Taft et al. 2007). Moreover, there is a broadly consistent rise in the amount of non-protein-coding intergenic and intronic DNA with developmental complexity, a relationship that proves nothing but which suggests an association that can only be falsified by downward exceptions, of which there are none known (Taft et al. 2007; Liu et al., 2013).
In contrast to these uncertain indices, estimations and interpretations, the major fact to emerge from the ENCODE studies (Birney et al. 2007; Dunham et al. 2012) and their predecessors (Cheng et al. 2005; Carninci et al. 2005) is that the vast majority of the mammalian genome is differentially transcribed in precise cell-specific patterns (Mercer et al. 2008) to produce large numbers of intergenic, interlacing, antisense and intronic non-protein-coding RNAs, which show dynamic regulation in embryonal development (Dinger et al. 2008; Guttman et al. 2011; Ng et al. 2012), tissue differentiation (Sunwoo et al. 2009; Pang et al. 2009; Mercer et al. 2010; Askarian-Amiri et al. 2011) and disease (Gupta et al. 2010; Khaitan et al. 2011), with even regions superficially described as ‘gene deserts’ expressing specific transcripts in particular cells (Mercer et al. 2012; Roberts and Pachter 2011). Moreover, there is increasing evidence of their functional relevance (Mattick 2009b) and that a major function of these noncoding RNAs is to guide chromatin-modifying complexes to their sites of action, to supervise the epigenetic trajectories of development (Mattick and Gagen 2001; Dinger et al. 2008; Nagano et al. 2008; Pandey et al. 2008; Khalil et al. 2009; Mattick et al. 2009; Koziol and Rinn 2010; Spitale et al. 2011) – which appears to comprise a far greater fraction of human genetic programming than expected (Mattick 2004) in order to specify the architecture of the organism at a level of detail well beyond mere cell-type specification (Mattick et al. 2010).
Continued Strong Resistance Among Most Scientists:
So, why such a strong resistance to this conclusion? – from many within the scientific community?
Unique Brain Development:
The uniqueness of the human brain, how it is formed, is based on ERV control. A gene regulatory network based on ERVs that participates in control of gene expression of protein-coding transcripts important for brain development has recently been discovered. “Several recent studies suggest that transposable elements (TEs), which make up more than one-half of the human DNA, have the capacity to establish primate-specific gene regulatory networks.” (Per Ludvik Brattås, 2017).
TEs are mobile genetic sequences with potential to alter the genetic landscape and to influence gene expression from their integration sites in the host genome by providing various cis-regulatory elements including promoters, repressors, enhancers, and insulators. It is becoming increasingly clear that TEs are adapted to influence gene expression through their regulatory sequences and that they play important roles in controlling and fine-tuning host gene networks (Cowley and Oakey, 2013).
Neurogenesis, in particular, involves a great many ERVs and other viral-type elements. Many different ERV loci are expressed (25,733 showing more than five reads/element in at least one sample) and demonstrate a dynamic expression across samples. Similarly, we found that other classes of TEs, such as LINE-1, Alu, and SVA elements, were also highly expressed in the embryonic brain samples. This demonstrates a dynamic ERV expression during neurogenesis when progenitors differentiate into neurons. A similar pattern was also observed when performing a cluster analysis including SINE, LINE, and SVA, revealing a highly region- and stage-specific expression of all main groups of TEs in the embryonic brain… Together, these data demonstrate a high-level and dynamic expression of ERVs during human brain development, which is controlled independently from protein-coding genes at different developmental stages. The data also demonstrate that the transcriptional control of ERVs in the developing brain is markedly different when compared to pluripotent stem cells resulting in the expression of primarily ERV fragments in the human embryonic CNS. This suggests the existence of a dynamic transcriptional repressor system with the capacity to induce local heterochromatin on and around ERVs at different stages during human brain development. (Per Ludvik Brattås, 2017).
Embryogenesis:
ERVs also play a key role in human embryogenesis in general – beyond brain development.
Pre-implantation embryo development encompasses several key developmental events, especially the activation of zygotic genome activation (ZGA)-related genes.
Endogenous retroviruses (ERVs), which are regarded as “deleterious genomic parasites”, were previously considered to be “junk DNA”. However, it is now known that ERVs, with limited conservatism across species, mediate conserved developmental processes (e.g., ZGA).
Transcriptional activation of ERVs occurs during the transition from maternal control to zygotic genome control, signifying ZGA. ERVs are versatile participants in rewiring gene expression networks during epigenetic reprogramming. Particularly, a subtle balance exists between ERV activation and ERV repression in host–virus interplay, which leads to stage-specific ERV expression during pre-implantation embryo development. A large portion of somatic cell nuclear transfer (SCNT) embryos display developmental arrest and ZGA failure during pre-implantation embryo development…
ERVs, as regulators of gene networks, play multiple critical roles during ZGA. In host–virus interplay, ERV activation is controlled by a multilayered regulatory network that maintains a balance between ERV activation and ERV repression, which results in stage-specific ERV expression during pre-implantation embryo development.
ERV-derived long noncoding RNAs (lncRNAs) participate in the control of pluripotency. Biomarkers of pluripotency, such as Oct4, Sox2, and Nanog, promote the expression of ESC-specific genes and suppress differentiation. In posttranscriptional networks, microRNAs (miRNAs) act as posttranscriptional modifiers and contribute to restraining pluripotency…. Particularly, many stem cell-specific transcription start sites are not associated with protein-coding genes, but with these kinds of ERV elements in mice and humans.
How exactly species-specific ERVs are activated to function as gene regulators in host cells and, conversely, how host cells defend themselves against ERV activation during the window of epigenetic reprogramming (pre-implantation embryo development) to prevent widespread retrotransposition, has been investigated. ERV transcripts are under acute surveillance by multilayered and interleaved systems that ensure a subtle balance between ERV activation and ERV repression, resulting in stage-specific ERV expression during pre-implantation embryo development. (Bo Fu, et. al., 2019)
Blocking other Viral Infections:
ERVs act to prevent disruptive viral infections.
The activation of ERVs may contribute to genome defense. LTR5HS, a subclass of HERV-K, is
transcribed from an LTR at the 8-cell stage (ZGA in human embryos) and contains the Oct4-binding
motif. Grow et al. [35] found that by binding to LTR5HS, Oct4 drove the expression of HERV-K
proviruses, producing viral-like particles and Gag proteins in pre-implantation embryos. Then,
the overexpression of Rec, HERV-K accessory protein, is sufficient to increase the level of virus
restriction factors such as IFITM1 during this process and contributes to fighting against exogenous
viral infections. (Bo Fu, et. al., 2019)
Placental Development:
Human placental development is also dependent upon ERVs.
One of the most iconic examples of retrovirus “domestication” is the gene Syncytin-1, which originates from a retroviral envelope gene. In primates, Syncytin-1 [and Syncytin-2] was repurposed for the development of a multinucleate tissue layer known as the syncytiotrophoblast, which separates maternal and fetal bloodstreams in the placenta. Remarkably, Syncytin-like retroviral proteins have been reported to be expressed in the placentas of nearly all mammals, yet Syncytins in different lineages derive from at least 10 independent infections by unrelated retroviruses. These findings have led to speculation that the co-option of unrelated ERVs in different species was a driving force underlying the evolutionary diversification of the placenta…
Functional studies have revealed that ERV-derived proteins tend to be co-opted for three roles in the placenta: mediating cell–cell fusion to form a multinucleate barrier, suppressing maternal immunity, and protecting the fetus from exogenous viruses. However, recent evidence suggests that ERVs may play an even more pervasive role in placenta evolution as noncoding regulatory elements (Chuong, 2018).
Different Types of Placentas for Different Functions:
Different placenta types needed for different animals with different gestational time requirements.
Despite having this critical and fundamentally similar function in all mammals, the placenta exhibits striking morphological variation (Mossman 1987; Leiser and Kaufmann 1994)… Commenting on the lack of any obvious pattern in the phylogenetic distribution of placental types, Benirschke and Kauffman (2006, p. 39) remark that this “may even give us the impression that several animals have acquired their respective placental types by chance.” Natural selection is unlikely to have been so permissive, however…
Placental invasiveness is the trait that has received the most attention regarding a possible impact of placental morphology on offspring prenatal development and maternal investment, particularly in relation to brain growth (Kihlstro¨m 1972; Sacher and Staffeldt 1974; Leutenegger 1979; Haig 1993; Crespi and Semeniuk 2004; Elliot and Crespi 2008; Martin 2008). (Capellini, et. al., 2011)
It is generally recognized that there are additional driving forces promoting adaptive changes in the placenta and hence contributing to its rapid evolution. One is undoubtedly the struggle for control of maternal physiology in terms of nutrient allocation (Fowden and Constancia 2012). As the demands of the fetus increase, they will likely conflict with the ability of the mother to provide such resources. An extensive literature has developed around this topic, with particular emphasis on the role played by imprinted genes in controlling nutrient supply and growth of the fetus (Reik, et al. 2003). (Roberts, et. al., 2016).
Different ERV sequences control placental development in different mammalian species that produce a placenta. All mammals except the egg-laying platypus and the five species of echidnas, the only surviving monotremes, rely on a placenta for their reproduction.
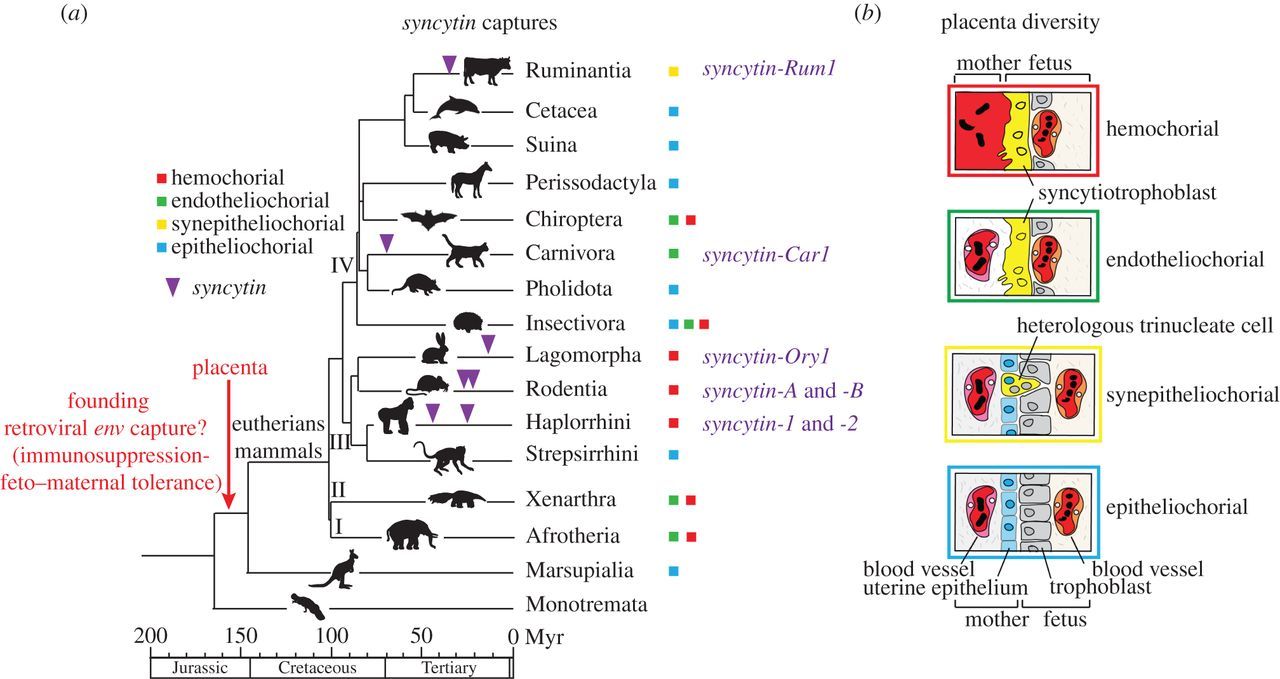
Although they share similar features, the primate and muroid syncytins are clearly not orthologous genes—they are not syntenic in the two species clades—indicating that they are the result of independent gene captures that occurred separately in the genome from ancestors of each lineage. Furthermore, a fifth syncytin gene, syncytin-Ory1, distinct from each of those just listed, has been identified in yet another mammalian lineage, the Leporidae family (rabbit and hare). (Christian Lavialle, 2013).
Control of Pregnancy and Birth Timing:
The control of human pregnancy and birth timing is based on ERVs.
Most research in this area has focused on ERV-derived proteins, which have been repeatedly co-opted to promote cell–cell fusion and immune modulation in the placenta. ERVs also harbor regulatory sequences that can potentially control placental gene expression, but there has been limited evidence to support this role. In a recent study, Dunn-Fletcher and colleagues discover a striking example of an ERV-derived enhancer element that has been co-opted to regulate a gene important for human pregnancy. Using genomic and experimental approaches, they firmly establish that a primate-specific ERV functions as a placenta-specific enhancer for corticotropin-releasing hormone (CRH), a hormone linked to the control of birth timing in humans. Their findings implicate an extensive yet understudied role for retroviruses in shaping the evolution of placental gene regulatory networks. (Chuong, 2018).
Intron Gains found almost Exclusively in Placental Mammals:
A significant amount of intron gain was found only in domesticated genes of placental mammals, where more than 70 cases were identified. De novo gained introns show clear positional bias, since they are distributed mainly in 5′ UTR and coding regions, while 3′ UTR introns are very rare. In the coding regions of some domesticated genes up to 8 de novo gained introns have been found. Intron densities in Eutheria-specific domesticated genes and in older domesticated genes that originated early in vertebrates are lower than those for normal mammalian and vertebrate genes. Surprisingly, the majority of intron gains have occurred in the ancestor of placentals. (Dušan Kordiš, 2011).
Greater Muscle Mass in Males:
Recently, researchers found that one of these viral proteins essential to placental development (syncytins), also increased fusion of myoblast cells during muscle-fiber formation: male mice lacking this retroviral gene for syncytin showed a 20% reduction in muscle mass.
Remarkably, this reduction is only observed in males, which subsequently show muscle quantitative traits more similar to those of females. In addition, we show that syncytins also contribute to muscle repair after cardiotoxin-induced injury, with again a male-specific effect on the rate and extent of regeneration.” (François Redelsperger, et. al., 2016) .
Most ERVs are Species Specific:
ERVs are usually species-specific… (RationalWiki, 2019)
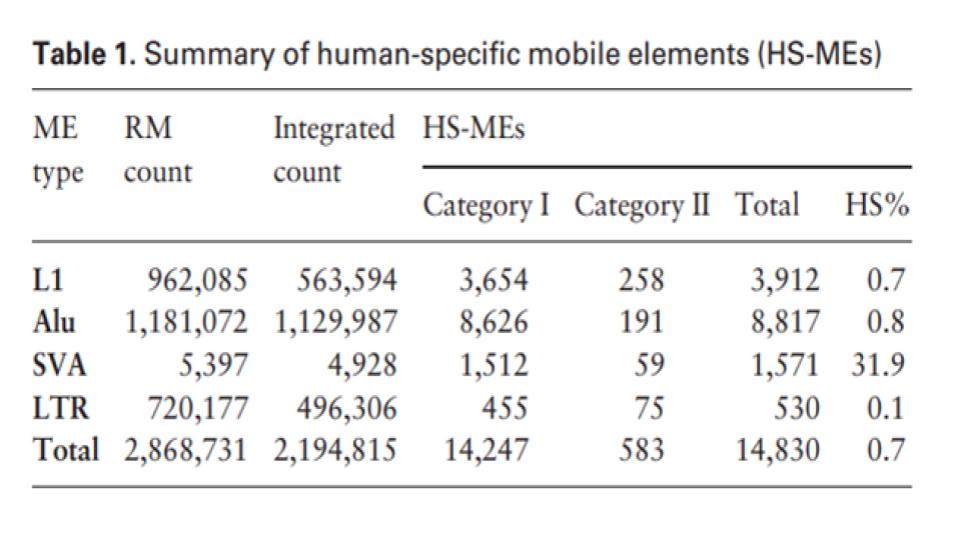
This isn’t exactly true. Consider, for example, that of the ~200,000 ERVs that are found in humans, the vast majority of these are also found in chimps. Of these, a 2001 study showed that 279 ERVs are specific to chimps while 82 are specific to humans (Nature, 2001). This finding was subsequently corroborated with studies showing less than 0.1% of ERVs to be lineage-specific (Polavarapu, Bowen, & McDonald, 2006) and then by a genome-wide comparison where virtually all ERVs were directly observed to be in identical loci (Chimpanzee Sequencing and Analysis Consortium, 2005).
Of course, since this time, thousands of additional human specific viral-type elements have been discovered (Tang, 2019). However, the overall ratio heavily favors matching ERVs between humans and chimps.
Beyond this, however, there are significant retroviral similarities between very different species – thought to be the result of common “horizontal transmission” between these species.
Notably, our results suggest a history of frequent horizontal transmission of gammaretroviruses and associated class I retroviral sequences during evolution… The phylogenetic pattern for ERVs from different host genomes suggests a striking mode of evolution in which interorder transmission—for example, between primates and rodents—is common. Here we show that this pattern may represent the default mode of evolution for the gammaretroviruses, because retroviral sequences from diverse mammals repeatedly occur adjacently in our phylogeny. These results imply an inherent capacity for gammaretroviruses to switch across diverse mammalian hosts…
An intriguing finding is the occurrence of cetacean sequences [killer whales and dolphins], in a highly supported clade, otherwise composed solely of mouse and rat ERVs… Our results thus suggest that cetaceans may have been exposed to this lineage as a consequence of rodent retrovirus transmission, perhaps via an unknown intermediate vector…
Our analyses suggest that a bottlenose dolphin (Tursiops truncatus) ERV… clusters with high support with cat and rabbit sequences at the base of the gammaretroviral clade. (Alexander Hayward, et. al., 2013).
Most Retroviruses Insert Randomly within a Genome:
ERVs… insert almost randomly in the host genome. (RationalWiki, 2019)
These seems like a reasonable conclusion given the hypothesis that all endogenous ERV sequences within a given genome where the result of a retroviral infection at some point in the past. However, given this assumption, it is interesting to note that ERV-type sequences are not distributed randomly within the host genome, but are clustered in “hot spot” regions.
But although this concept of retrovirus selectivity is currently prevailing, practically all genomic regions were reported to be used as primary integration targets, however, with different preferences. There were identified ‘hot spots’ containing integration sites used up to 280 times more frequently than predicted mathematically. (Sverdlov, 2000)
Moreover, for DNA transposons, it has been proposed that recombination hotspots are required by the transposition mechanism, and perhaps a similar interaction is essential for ERVs. (Rebeca Campos-Sánchez, et. al., 2014)
We found that fixed mouse ERVs are located closer to centromeres than polymorphic mouse ERVs, and that the latter are located closer to centromeres than control regions. The preferential location of ERVs next to centromeres might be explained by their integration in AT-rich regions and by their fixation in regions with low recombination rates (Rebeca Campos-Sánchez, et. al., 2016)
Vertebrate retrotransposons present at specific loci have been deemed to be essentially homoplasy-free phylogenetic characters because the probability of insertion occurring more than once at any single site has been presumed to be vanishingly low (Batzer and Deininger 1991; Batzeret et. al., 1994; Takahashiet et al. 1998; Nikaidoet et al. 1999)… Vertebrate retrotransposons have been used extensively for phylogenetic analyses and studies of molecular evolution. Information can be obtained from specific inserts either by comparing sequence differences that have accumulated over time in orthologous copies of that insert or by determining the presence or absence of that specific element at a particular site. The presence of specific copies has been deemed to be an essentially homoplasy-free phylogenetic character because the probability of multiple independent insertions into any one site has been believed to be nil. . . . We have identified two hot spots for SINE insertion within mys-9 and at each hot spot have found that two independent SINE insertions have occurred at identical sites. These results have major repercussions for phylogenetic analyses based on SINE insertions, indicating the need for caution when one concludes that the existence of a SINE at a specific locus in multiple individuals is indicative of common ancestry. Although independent insertions at the same locus may be rare, SINE insertions are not homoplasy-free phylogenetic markers. (Cantrell, et al, 2001, 158:769)
The insect retrotransposons R1 and R2 limit insertion almost exclusively to the 28S rRNA genes (Xiong and Eickbush 1988; Baker and Wichman 1990; Sandmeyeret et al. 1990; Wichmanet et al. 1992; Craig 1997).
Slattery and co-workers found what appear to be two independent insertions of the same tRNA-derived SINE element at the same site in members of the cat family (Felis silvestris and Lynx rufus; Slatteryet et al. 2000).
A clear example of two independent B1 inserts into the same site in the genus Mus has recently been found by Kass and co-workers (2000).
Consider also the comments on this topic from Dr. Anjeanette Roberts, a research scholar in molecular biology and virology:
Despite early findings in vitro, retroviral insertion sites are not selected randomly. Various retroviruses have varying degrees of insertion site preferences, some showing site bias and others even demonstrating integration specificity at the primary sequence level. There are a variety of factors now known to effect integration site specificity. These include different viral proteins (IN, Gag, U3 LTR), chromatin accessibility (A/T-rich distorted DNA and outwardly facing major grooves), cell-cycle effects (integration in dividing cells occurs at a much higher rate, and increased site specificity is observed in integration in non-dividing cells), and cellular integration co-factors (tethering proteins like LEDGF/p75, gene regulatory elements, and epigenetic marks). Since a range of insertion site specificities and contributing factors exist for the various classes of retroviruses, it is possible that retroviral infections establishing the shared ERV sites in NHP chromosomal segments had even greater specificity for insertion site selection than those actively tested and observed to date…
Another observation of shared NHP ERVs that is contrary to evolutionary predictions involves the divergence of sequences in paired 5’ and 3’ proviral LTRs, which accrue differing mutations at similar rates following insertion. Divergence between LTR sequences at a single shared ERV site sometimes varies more significantly in one species than in another, suggesting differences in times of insertion between the two species. For example, estimated divergence of chimpanzee 1p31.1a proviral LTRs is 6.5 times greater than that observed in humans. Human 1p31.1a is also dimorphic with solo LTR and provirus, unlike the chimp ortholog. Both of these findings suggest a much more recent integration event in humans than in chimps at orthologous sites.
Although evolutionary arguments are made to account for these observations, independent ERV infection events with similar insertion site specificities offer simpler viable explanations for ERVs that do not track with phylogenetic predictions based on NHP species relatedness. (Anjeanette Roberts, 2015)
Of course, this isn’t specific enough to entirely account for the degree of consistency of matching ERVs between humans and apes. Rather, given the high level of functionality that is being discovered for ERVs, it is much more consistent to attribute this degree of functionality to deliberate design. It would therefore follow that those creatures with similar functionality would also have similar ERVs in similar locations within their respective genomes. In other words, ERVs could exist in similar places in different genomes with the same or a similar sequence in order to perform an essential function common to very similar organisms.
A Nested Hierarchical Pattern:
Inconsistent Patterns:
When phylogenetic trees are constructed based on the pattern of ERVs, they indicate humans share more ERVs with chimps than either share with gorillas. (RationalWiki, 2019)
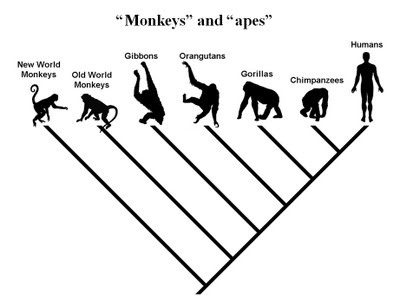
What is also interesting, given this particular argument, is that the expected phylogenies don’t always work. For example, humans and chimps are thought to be more closely related to their most recent common ancestor (MRCA) than are humans and gorillas. Yet, there are some features of the genomes that indicate a closer relationship between chimps and gorillas, as compared to humans.
We identified a human endogenous retrovirus K (HERV-K) provirus that is present at the orthologous position in the gorilla and chimpanzee genomes, but not in the human genome. Humans contain an intact preintegration site at this locus. (Barbulescu, 2001)
How is this explained? Well, it’s explained through a fairly convoluted argument where portions of the human genome are more closely related to chimps while other portions are more closely related to gorillas.
These observations provide very strong evidence that, for some fraction of the genome, chimpanzees, bonobos, and gorillas are more closely related to each other than they are to humans… (Barbulescu, 2001)
So, the phylogenies of some parts of a genome can show one type of evolutionary ancestry while the phylogenies based on other parts of the same genome can show a very different evolutionary relationship and common ancestry? Seems rather convenient does it not?
Though there are other possible candidate hypotheses for this observation (such as incomplete lineage sorting), in the context of other indications of locus-specific site preference, this data suggests, from the evolutionary perspective at least, that these inserts could have been the result of independent events.
Consider also that in the water flea genome introns are routinely found at the very same loci without an evident evolutionary relationship or common ancestry (Li, et. al., 2009).
Remarkably, we have found many cases of parallel intron gains at essentially the same sites in independent genotypes. This strongly argues against the common assumption that when two species share introns at the same site, it is always due to inheritance from a common ancestor. (Lynch, 2009)
Environmental Influence:
Strikingly, we find that six large vegetarian mammals (cattle, llama, horse, panda, sloth, and elephant) possess multiple class I ERVs, but no representative gamma-ERVs. Additionally, in our screening, the vegetarian wallaby has only a single gamma-ERV, and the manatee—which feeds on marine plants, fish, and invertebrates—also harbors no gamma-ERVs. Conversely, the only true carnivores included in our analysis (the cat, tarsier, and, to some extent, the dog) contain an overrepresentation of gamma-ERVs relative to other class I ERVs… This suggestion is similar to arguments regarding murine-to-porcine transmission based on shared environments between pig and mouse, invoked to explain the close relationship observed between MLVs and PERVs. Although other vegetarian host taxa possess gamma-ERVs (gorilla, orangutan, guinea pig, mole rat, pika, rabbit, and megabat), all these host taxa, with the exception of megabat, belong to the Euarchontoglires, which is the taxonomic group containing the highest overall representation of gamma-ERVs. It may be possible that these euarchontoglire hosts are not strictly vegetarian because recent studies suggest that several of these taxa do occasionally eat meat, given that even rare carnivorous acts may be sufficient to expose hosts to retroviral infection (Alexander Hayward, et. al., 2013).
It seems, then, that some ERV patterns may be more related to environmental conditions rather than common descent… which would affect the nature of the nested hierarchical patterns for these genetic sequences.
False Patterns due to Convergence:
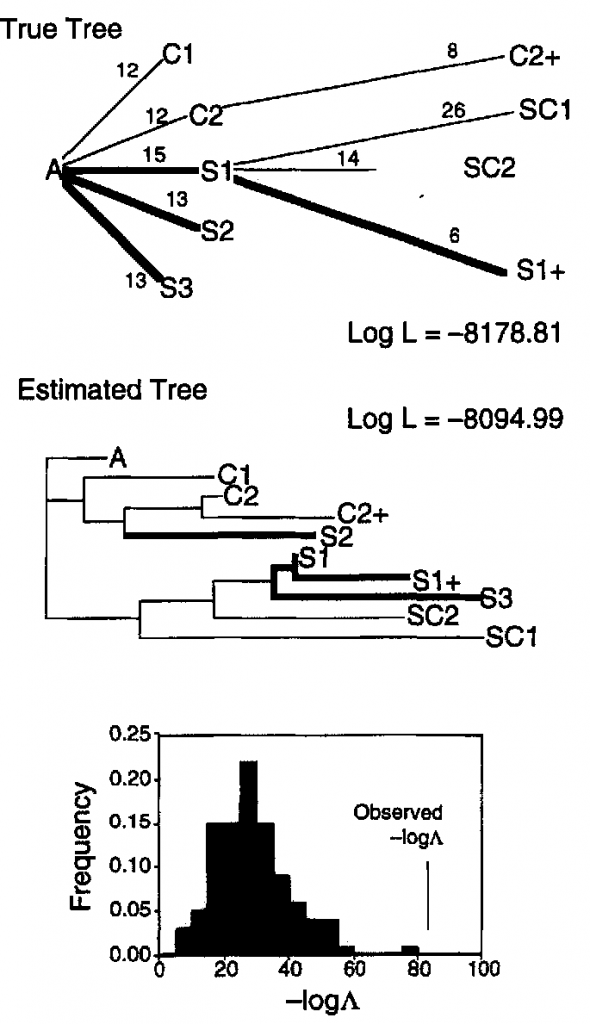 Another interesting example of this phenomenon has been studied in detail in more rapidly reproducing organisms, such as viruses. For example, an interesting study was published by Bull et al. (1997) on replicate lineages of the bacteriophage phiX174. Numerous mutations occurred in each genome during propagation. Across nine separate lineages 119 independent substitutions occurred at 68 nucleotide sites. What is interesting here is that over half of these substitutions at 1/3 of the sites were identical in the different lineages. Some convergent substitutions were specific to specific hosts while others where shared between the two separate hosts.
Another interesting example of this phenomenon has been studied in detail in more rapidly reproducing organisms, such as viruses. For example, an interesting study was published by Bull et al. (1997) on replicate lineages of the bacteriophage phiX174. Numerous mutations occurred in each genome during propagation. Across nine separate lineages 119 independent substitutions occurred at 68 nucleotide sites. What is interesting here is that over half of these substitutions at 1/3 of the sites were identical in the different lineages. Some convergent substitutions were specific to specific hosts while others where shared between the two separate hosts.
Phylogenetic reconstruction using the complete genome sequence not only failed to recover the correct evolutionary history because of these convergent changes, but the true history was rejected as being a significantly inferior fit to the data.
In a subsequent similar study Bull et al. argue that such results “point to a limited number of pathways taken during evolution in these viruses, and also raise the possibility that much of the amino-acid variation in the natural evolution of these viruses has been selected.” (Bull et. al., 2000). In other words, much of the variations in viral genomes is not neutral, but is in fact functional and therefore maintained by natural selection.
This is amazing! The implications here are quite stunning. If the convergent nature of molecular mutations like this cannot be adequately detected such mutations would interfere with any sort of reliable phylogenetic tree building or accurate determination of evolutionary relationships. If there is any sort of correlation with higher-level multicellular organisms, this could significantly undermine the entire science of evolutionary biology as it is currently understood. Real time studies like this are obviously needed on a wider scale to determine if such convergent mutations are more widespread. Obviously, the common assumption that convergent mutations on the molecular level are rare and the result of completely random chance is simply not true anymore for at least some (and possibly most if not all) genomes.
A similar finding was described more recently by Cuevas et al. in a 2002 article published in Genetics dealing with RNA viruses (Link). In this study the authors again demonstrated convergences in 12 variable sites in independent lineages. The authors were surprised to discover that convergences occurred not only within non-synonymous sites, but in synonymous sites and intergenetic regions as well (usually thought to be neutral with respect to the effects of natural selection). The authors also noted that this phenomenon is not restricted to the laboratory, but is also a relatively widespread observation among HIV-1 virus clones in humans and in SHIV strains isolated from macaques, monkeys, and humans. These same authors go on to note:
Convergent evolution at the molecular level is not controversial as long as it can be reconciled with the neutralist and the selectionist theories. The neutral theory suggests that convergences are simply accidents, whereas within the framework of selectionism, there are two qualifications for convergences. The first explanation considers convergences as being adaptive and the result of organisms facing the same environment (as in the case of our experiments) with a few alternative pathways of adaptation (as expected for compacted genomes). Second, keeping in mind the model of clonal interference, beneficial mutations have to become fixed in an orderly way (Gerrish and Lenski 1998), with the best possible candidate fixed first, and then the second best candidate, and so on. This implies that, given a large enough population size to make clonal interference an important evolutionary factor, we should always expect the same mutations to be fixed. (Cuevas et al., 2002)
According to the authors, the the above argument is valid for nonsynonymous changes but an alternative explanation must be found for synonymous changes and for changes in the intergenic regions since these changes are generally though to be selectively neutral. So, the authors note:
Genomic RNA is involved in many RNA-RNA and RNA-protein interactions that affect viral replication. This is obvious for noncoding, regulatory regions (Stillman and Whitt 1997, 1998), but there is increasing evidence that capsid-coding regions in picornaviruses may also have an effect on viral replication (McKnight and Lemon 1998; Fares et al. 2001). Therefore, the RNA itself (apart from its protein-coding capacity) may contribute to the viral phenotype, and fitness may also be affected by synonymous replacements.” This is an important point because, “Evidence for selection on synonymous sites has been inferred also in mammals (Eyre-Walker 1999), as a consequence of selection acting upon the base composition of isochors and large sections of junk DNA. (Cuevas et al., 2002)
In other words, there doesn’t seem to be much DNA, even in seemingly non-functional areas of DNA or even among synonymous changes, that is truly non-functional when it comes to viral genomes. The authors then go on to suggest a comparison with the genomes of high-level organism, like hominids.
For example, Fay et al (2001) reported that, in humans, the vast majority (80%) of amino acidic changes are deleterious to some extent and only a minor fraction are neutral. Among these deleterious amino acidic mutations, at least 20% are slightly deleterious. Here, we found that 15 amino acid sites changed, with only 5 being significantly advantageous. At this point, we can only speculate about the selective role of all the amino acid sites shown to be invariable in our study. The total number of amino acids in five genes of VSV is 3536. Assuming that changes in any of the 3536 – 15 – 3521 invariable amino acids would be deleterious (and thus washed out by purifying selection during or evolution experiment), then the fraction of amino acid replacements that are potentially harmful would be 3521/3536 = ~99.58%; the fraction of neutral sites would be 10/3536 = ~0.28%; whereas only 5/3536 = ~0.14% would be beneficial. Despite the differences between humans and VSV in genome size and organization and in the nature of the nucleic acid used, in both cases the fraction of potentially deleterious amino acid substitutions is overwhelmingly larger than that of neutral or beneficial ones. (Cuevas et al., 2002)
In other words, it is at least reasonable to suspect that very little coding DNA, even in hominids, is truly “neutral” or immune to all pressures of natural selection. This is becoming true of non-coding DNA as well given that much of what was once thought to be junk is now being found to be functional (Link). This strongly suggests that many of what were thought to be shared mutational errors might actually be functionally-maintained by similar creatures in similar environments. In this light, consider the following conclusions of Wood et al published in a 2005 edition of Genetica:
The most convincing evidence of parallel genotypic adaptation comes from artificial selection experiments involving microbial populations. In some experiments, up to half of the nucleotide substitutions found in independent lineages under uniform selection are the same. Phylogenetic studies provide a means for studying parallel genotypic adaptation in non-experimental systems, but conclusive evidence may be difficult to obtain because homoplasy can arise for other reasons. Nonetheless, phylogenetic approaches have provided evidence of parallel genotypic adaptation across all taxonomic levels, not just microbes. Quantitative genetic approaches also suggest parallel genotypic evolution across both closely and distantly related taxa, but it is important to note that this approach cannot distinguish between parallel changes at homologous loci versus convergent changes at closely linked non-homologous loci. The finding that parallel genotypic adaptation appears to be frequent and occurs at all taxonomic levels has important implications for phylogenetic and evolutionary studies. With respect to phylogenetic analyses, parallel genotypic changes, if common, may result in faulty estimates of phylogenetic relationships. (Wood et. al., 2005)
Notice that according to Wood et al, parallel and/or convergent mutations are “frequent” at “all taxonomic levels, not just microbes”. That’s very interesting and does indeed have very serious implications when it comes to determining phylogenetic relationships – relationships that are likely to be not only wrong, but meaningless as far as the evolutionary theory of common descent is concerned. Rather, phylogenetic similarities may be more a reflection of functional similarities and differences than of true evolutionary relationships.
The error or mutation that inactivated the gene is random. (RationalWiki, 2019)
Regarding shared “mistakes” between primate genomes, this argument again assumes that mutations are generally random and are unlikely to occur convergently.
Evolutionary Convergence in Viruses:
Here, we report a full-genome sequence analysis of 21 independent populations of vesicular
stomatitis ribovirus evolved on the same cell type but under different demographic regimes. Each demographic regime differed in the effective viral population size. Evolutionary convergences are widespread both at synonymous and nonsynonymous replacements as well as in an intergenic region. (Cuevas et al., 2002),
Some of these convergent mutations even took place in intergenic regions (changes in which are normally thought to be selectively neutral) and also in synonymous sites. The authors also note that this observation is fairly widespread among HIV-1 virus clones in humans and in SHIV strains isolated from macaques, monkeys and humans. As the authors note:
One of the most amazing features illustrated in Figure 1 is the large amount of evolutionary convergences observed among independent lineages. Twelve of the variable sites were shared by different lineages. More surprisingly, convergences also occurred within synonymous sites and intergenic regions. Evolutionary convergences during the adaptation of viral lineages under identical artificial environmental conditions have been described previously (Bull et al. 1997; Wichman et al. 1999; Fares et al. 2001). However, this phenomenon is observed not only in the laboratory. It is also a relatively widespread observation among human immunodeficiency virus (HIV)-1 clones isolated from patients treated with different antiviral drugs; parallel changes are frequent, often following a common order of appearance (Larder et al. 1991; Boucher et al. 1992; Kellam et al. 1994; Condra et al. 1996; Martinez-Picado et al. 2000). Subsequent substitutions may confer increasing levels of drug resistance or, alternatively, may compensate for deleterious pleiotropic effects of earlier mutations (Molla et al. 1996; Martinez-Picado et al. 1999; Nijhuis et al. 1999). Also, molecular convergences have been observed between chimeric simian-human immunodeficiency viruses (strain SHIV-vpu+) isolated from pig-tailed macaques, rhesus monkeys, and humans after either chronic infections or rapid virus passage (Hofmann-Lehmann et al. 2002). (Cuevas et al., 2002)
Convergence in Pseudogenes:
Similar “shared mistakes” are also common within “pseudogenes”. Mutational “hotspots” have been identified in many genes as well as pseudogenes (Pitman, 2016).
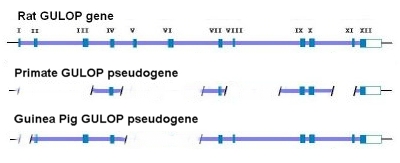 Consider the GULOP (or GULO) pseudogene for example. In most mammals this is an active gene encoding the enzyme L-glucono-γ-lactone oxidase (LGGLO). GULO is located on chromosome 8 at p21.1 in a region that is rich in genes (see figure). This is the enzyme that catalyzes the last step in the synthesis of ascorbic acid (vitamin C). As it turns out, this particular gene is defective in humans and other primates as well as several other creatures to include guinea pigs, bats and certain kinds of fish. Compared to the rat GULO gene, the human version, as well as the great ape version, has large or clearly functional deletions involving exons I-III, V-VI, VIII, and XI (see figure above). Compare this with the significant deletions of the guinea pig GULO sequence that involve exons I, V, and VI – – all of which match the same losses of the primate mutations. In addition to this, all four functionally detrimental stop codons (3TGA and 1TAA sequences) that are identified in the guinea pig are shared at the same sites locations in the primate GULO pseudogene.
Consider the GULOP (or GULO) pseudogene for example. In most mammals this is an active gene encoding the enzyme L-glucono-γ-lactone oxidase (LGGLO). GULO is located on chromosome 8 at p21.1 in a region that is rich in genes (see figure). This is the enzyme that catalyzes the last step in the synthesis of ascorbic acid (vitamin C). As it turns out, this particular gene is defective in humans and other primates as well as several other creatures to include guinea pigs, bats and certain kinds of fish. Compared to the rat GULO gene, the human version, as well as the great ape version, has large or clearly functional deletions involving exons I-III, V-VI, VIII, and XI (see figure above). Compare this with the significant deletions of the guinea pig GULO sequence that involve exons I, V, and VI – – all of which match the same losses of the primate mutations. In addition to this, all four functionally detrimental stop codons (3TGA and 1TAA sequences) that are identified in the guinea pig are shared at the same sites locations in the primate GULO pseudogene.
Of course, it seems that we humans are able to get along just fine without this gene because we eat a lot of foods that are rich in vitamin C, like citrus fruits. So, what’s the big deal? Well, the argument goes something like this (as per a popular Talk.Origins essay by Edward E. Max, Ph.D.):
In most mammals functional GLO genes are present, inherited – according to the evolutionary hypothesis – from a functional GLO gene in a common ancestor of mammals. According to this view, GLO gene copies in the human and guinea pig lineages were inactivated by mutations. Presumably this occurred separately in guinea pig and primate ancestors whose natural diets were so rich in ascorbic acid that the absence of GLO enzyme activity was not a disadvantage–it did not cause selective pressure against the defective gene.
Molecular geneticists who examine DNA sequences from an evolutionary perspective know that large gene deletions are rare, so scientists expected that non-functional mutant GLO gene copies–known as “pseudogenes”–might still be present in primates and guinea pigs as relics of the functional ancestral gene. . . [Beyond this], the theory of evolution would make the strong prediction that primates [like apes and monkeys] would carry similar crippling mutations to the ones found in the human pseudogene. A test of this prediction has recently been reported. A small section of the GLO pseudogene sequence was recently compared from human, chimpanzee, macaque and orangutan; all four pseudogenes were found to share a common crippling single nucleotide deletion that would cause the remainder of the protein to be translated in the wrong triplet reading frame (Ohta and Nishikimi BBA 1472:408, 1999).
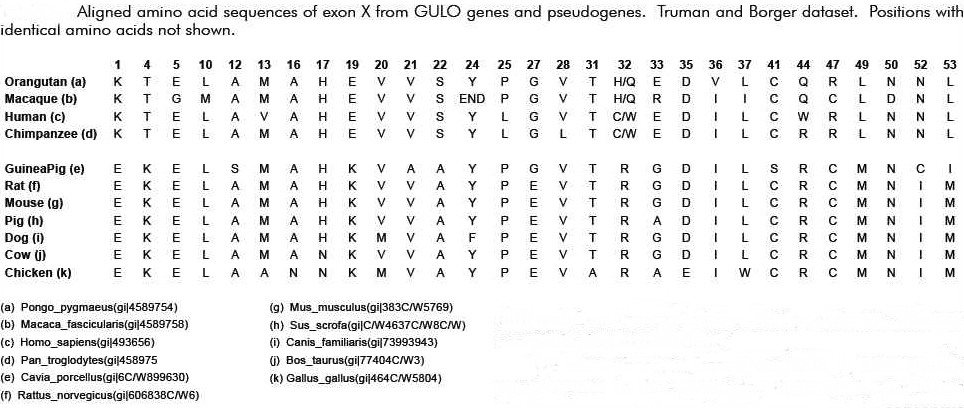
Now, it is interesting that among the many various substitution mutations in the “GLO” pseudogene that many, though not all, would be shared, to include a single deletion mutation that is shared by all primates (when compared to the rat of course). If not for common descent why would the sequences of human, chimpanzee, gorilla and orangutan reveal a single nucleotide deletion at position 97 in the coding region of Exon X? What are the odds that out of 165 base pairs the same one would be mutated in all these primates by random chance? Pretty slim – right? Is this not then overwhelming evidence of common evolutionary ancestry?
This would indeed seem to be the case at first approximation. However, in 2003, the same Japanese group published the complete sequence of the guinea pig GLO pseudogene, which is thought to have evolved independently, and compared it to that of humans [Inai et al, 2003]. 21 Surprisingly, they reported many shared mutations (deletions and substitutions) present in both humans and guinea pigs. Remember now that humans and guinea pigs are thought to have diverged at the time of the common ancestor with rodents. Therefore, a mutational difference between a guinea pig and a rat should not be shared by humans with better than random odds. But, this was not what was observed. Many mutational differences were shared by humans, including the one at position 97. According to Inai et al, this indicated some form of non-random bias that was independent of common descent or evolutionary ancestry. The probability of the same substitutions in both humans and guinea pigs occurring at the observed number of positions was calculated, by Inai et al, to be 1.84×10-12 – consistent with mutational hotspots.

What is interesting here is that the mutational hot spots found in guinea pigs and humans exactly match the mutations that set humans and primates apart from the rat (see figure below). This particular feature has given rise to the obvious argument that Inai et al got it wrong. Reed Cartwright, a population geneticist, has noted a methodological flaw in the Inai paper:
“However, the sections quoted from Inai et al. (2003) suffer from a major methodological error; they failed to consider that substitutions could have occurred in the rat lineage after the splits from the other two. The researchers actually clustered substitutions that are specific to the rat lineage with separate substitutions shared by guinea pigs and humans. . .
If I performed the same analysis as Inai et al. (2003), I would conclude that there are ten positions where humans and guinea pigs experienced separate substitutions of the same nucleotide, otherwise known as shared, derived traits. These positions are 1, 22, 31, 58, 79, 81, 97, 100, 109, 157. However, most of these are shown to be substitutions in the rat lineage when we look at larger samples of species.
When we look at this larger data table, only one position of the ten, 81, stands out as a possible case of a shared derived trait, one position, 97, is inconclusive, and the other eight positions are more than likely shared ancestral sites. With this additional phylogenetic information, I have shown that the “hot spots” Inai et al. (2003) found are not well supported.” (Link)

It does indeed seem like a number of the sequence differences noted by Cartwright are fairly unique to the rat – especially when one includes several other species in the comparison. However, I do have a question regarding this point. It seems to me that there simply are too many loci where the rat is the only odd sequence out in Exon X (i.e., there are seven and arguably eight of these loci). Given the published estimate on mutation rates (Drake) of about 2 x 10-10 per loci per generation, one should expect to see only 1 or 2 mutations in the 164 nucleotide exon in question (Exon X) over the course of the assumed time of some 30 Ma (million years). Therefore, the argument of the mutational differences being due to mutations in the rat lineage pre-supposes a much greater mutation rate in the rat than in the guinea pig. The same thing is true if one compares the rat with the mouse (i.e., the rat’s evident mutation rate is much higher than that of the mouse).
This is especially interesting since many of the DNA mutations are synonymous (Link). Why should essentially neutral mutations become fixed to a much greater extent in the rat gene pool as compared to the other gene pools? Wouldn’t this significant mutation rate difference, by itself, seem to suggest a mutationally “hot” region – at least in the rat?
Beyond this, several loci differences are not exclusive to the rat/mouse gene pools and therefore suggest mutational hotspots beyond the general overall “hotness” or propensity for mutations in this particular genetic sequence.
Some have noted that although the shared mutations may be the result of hotspots, there are many more mutational differences between humans and rats/guinea pigs as compared to apes. Therefore, regardless of hotspots, humans and apes are clearly more closely related than are humans and rats/guinea pigs.
The problem with this argument is that the rate at which mutations occur is related to the average generation time. Those creatures that have a shorter generation time have a correspondingly higher mutation rate over the same absolute period of time – like 100 years. Therefore, it is only to be expected that those creatures with relatively long generation times, like humans and apes, would have fewer mutational differences relative to each other over the same period of time relative to those creatures with much shorter generation times – like rats and guinea pigs.
What is interesting about many of these mutational losses is that they often share the same mutational changes. It is at least reasonably plausible then that the GULO mutation could also be the result of similar genetic instability that is shared by similar creatures (such as humans and the great apes).
This same sort of thing is seen to a fairly significant degree in the GULO region. Many of the same regional mutations are shared between humans and guinea pigs. Consider the following illustration yet again:
Why would both humans and guinea pigs share major deletions of exons I, V and VI as well as four stop codons if these mutations were truly random? In addition to this, a mutant group of Danish pigs have also been found to show a loss of GULO functionality. And, guess what, the key mutation in these pigs was a loss of a sizable portion of exon VIII. This loss also matches the loss of primate exon VIII. In addition, there is a frame shift in intron 8 which results in a loss of correct coding for exons. This also reflects a very similar loss in this region in primates (Link). That’s quite a few key similarities that were clearly not the result of common ancestry for the GULO region. This seems to be very good evidence that many if not all of the mutations of the GULO region are indeed the result of similar genetic instabilities that are prone to similar mutations – especially in similar animals.
As an aside, many other genetic mutations that result in functional losses are known to commonly affect the same genetic loci in the same or similar manner outside of common descent. For example, achondroplasia is a spontaneous mutation in humans in about 85% of the cases. In humans achondroplasia is due to mutations in the FGFR2 gene. A remarkable observation on the FGFR2 gene is that the major part of the mutations are introduced at the same two spots (755 C->G and 755-757 CGC->TCT) independent of common descent. The short legs of the Dachshund are also due to the same mutation(s). The same allelic mutation has occurred in sheep as well.
Genomic Fixation:
Consider again that over half of the human genome is comprised of viral-type sequences. Assuming that the majority of these sequences were the result of a retroviral infection, how did they all get fixed within the ancestral germline of all humans?
When a new neutral mutation occurs in a constant population of size N that is undergoing random mating, the probability that it will ultimately become fixed is approximately ½N. For those mutations that do become fixed, the average time to fixation is approximately 4N generations. A detailed analysis of data on human genetic variation suggests an ancestral population size of approximately 10,000 during the period when the current pattern of genetic variation was largely established. Under these conditions, the probability that a new, selectively neutral mutation would become fixed was small (5 × 10–5), while the average time to fixation was on the order of 800,000 years. Thus, while we know that the human population has grown enormously since the development of agriculture approximately 15,000 years ago, most human genetic variation arose and became established in the human population much earlier than this, when the human population was still small. (Bruce Alberts, et. al., 2002)
So, assuming a very small ancestral population size of only 10,000 or so individuals on the planet, the average time for a novel genetic sequence, assuming functional neutrality, would be around 800,000 years. And, that’s if it ever got fixed at all. The odds that are particular sequence would become fixed is very small – at around 5 × 10–5 (or about 1 chance in 20,000 tries).
In this light, consider again that the human genome contains over 2 million viral-type sequences (Tang, 2019). This suggests 20,000 times the number of viral infections that made into the germline to begin with, but were never fixed – i.e., around 40 billion infestations to get to where we are now. Of course, the average fixation time of 800 kyrs is basically irrelevant given the assumption of tens of millions of years to work with. However, this does seem to beg the question as to why there are no known examples of retroviral endogenization into the human genome today? or any ape genome? – or any other known genome of any kind for that matter? – with the lone exception of the Koala Bear? (Stoye, 2006)
Add to this the fact that “modern human retroviruses are known to infect somatic cells, primarily of hematopoietic origin, not gametes or gametic precursors. Gametic cell-types apparently lack the appropriate receptors for viral entry. A second obstacle to proviral insertion is raised by the observation that retroviral DNAs of some retroviruses do not integrate into the DNA of quiescent (inactive), nonreplicating cells—such as gametes. Therefore, proviral integration into an ancient gametic chromosome faces two known obstacles: one at viral entry and another at proviral integration.” (Anjeanette Roberts, 2015)
Yet, if such events were downright commonplace in the past, despite these difficulties, for most animal species, what’s the reason for the dramatic lack of such examples today?
We speculate that the decline in ERV integration in the human genome has been exacerbated by a relatively low burden of horizontally-transmitted retroviruses and subsequent reduced risk of endogenization. (Gkikas Magiorkinis, 2015)
But that doesn’t really answer the question, does it? I mean, why is there suddenly a “relatively low burden of horizontally-transmitted retroviruses”? It’s like answering a question by posing another apparently inexplicable mystery or a just-so story.
Origin of ERVs and other Retroviruses:
Retroviruses are a virus family of considerable medical and veterinary importance. Until recently, very little was known about deep retroviral origins. New research supports a marine origin of retroviruses, ∼460–550 million years ago. The evolutionary events leading to the origin of retroviruses remain obscure. (Alexander Hayward, 2017)
In short, then, nobody really knows how informationally and functionally complex retroviruses evolved any more than anyone knows how the first living thing could have evolved via mindless natural mechanisms – or any biological system for that matter beyond the lowest levels of functional complexity (i.e., requiring more than a few hundred specifically-arranged nucleotides or amino acid residues). Statistically, such evolutionary progress would require trillions upon trillions of years of time making such a story rationally untenable (Link, Link). Beyond the lowest levels of functional complexity, the only mechanisms that are known to produce novel functionality require the involvement deliberate intelligent design at one level or another. Therefore, the only truly scientific conclusion regarding the origin of the complex structure and functionality of retroviruses is intelligent design as well.
This does not mean, of course, that all retroviruses currently function as they were originally designed to function. It is possible for good designs to break down over time via random insults. Devolution can be demonstrated at all levels of functional design in real time. However, achieving higher level functionality to begin with, cannot originate with random natural mechanisms of any kind.
Summary:
 Endogenous Retroviruses (ERVs) and other viral elements that are found within the genomes of a very wide variety of organisms would seem to pose a significant block of evidence in favor of the Theory of Evolution – particularly regarding the concept of the common descent of all organisms from a single common ancestor that came into existence on this planet a few billion years ago. After all, viruses are generally viewed in a very negative light since they are associated with a host of diseases and other problems. So, the discovery of such viral sequences in the same position within humans and other similar species (like apes), would seem to strongly suggest a common evolutionary ancestry. How else could such shared viral-type sequences be explained?
Endogenous Retroviruses (ERVs) and other viral elements that are found within the genomes of a very wide variety of organisms would seem to pose a significant block of evidence in favor of the Theory of Evolution – particularly regarding the concept of the common descent of all organisms from a single common ancestor that came into existence on this planet a few billion years ago. After all, viruses are generally viewed in a very negative light since they are associated with a host of diseases and other problems. So, the discovery of such viral sequences in the same position within humans and other similar species (like apes), would seem to strongly suggest a common evolutionary ancestry. How else could such shared viral-type sequences be explained?
What are the odds that a host of different viruses would integrate themselves within the genomes of both humans and apes, independently, at the very same positions? – by random chance? That notion is ridiculous! – right? Who, in their right mind, would believe that? Add to this the presence of the very same functionally-relevant mutations within the very same viral sequences and the explanatory power of the common descent hypothesis becomes downright overwhelming! – right? I mean, what else could explain these sorts of observations? – besides common descent from a shared common ancestor?
The proposal that an intelligent designer could be responsible for such a situation seems downright desperate, even laughable, at this point. The conclusion seems inescapable that those who continue to propose an intelligent designer, or God, as being somehow responsible, is necessitated only by religious motivations for those who have no other option to explain what is, otherwise, overwhelming evidence against this notion.
After all, what designer, in his or her right mind, would produce a nested hierarchical pattern (NHP) of viral-type infections within human and ape genomes (as well as a host of other species) that would be so predictable by a process of common descent? – rather than common design? Why not use a design that would undermine any rational conclusion for any other mechanism besides common design? As it is, what is there, really, that is left to favor the common design hypothesis over and above the otherwise obvious conclusion of common descent?

Well, I’ve learned over the years that things are not always as they might appear at first approximation. And, when it comes to a hierarchical pattern of matching ERVs within various genomes, it might be wise to step back a bit and consider a few additional facts and ask a few additional questions before making up one’s mind.
- The functional complexity of functional viruses and viral sequences is quite high. In fact, it is so high that no naturalistic mechanism (not even random mutations and function-based selection) can explain the origin of such functional complexity. Despite the grand claims of many scientists to the contrary, none of these claims are backed up by demonstration or anything that resembles good science. To the contrary, the actual scientific evidence that is in hand strongly speaks to a definitive limit to evolutionary progress at very low levels of functional complexity – much lower than that found in functional viruses and viral sequences (Link). Therefore, the evidence, as I understand it, overwhelmingly favors the conclusion that viruses where originally designed by an intelligent mind.
- If viruses were originally deliberately designed, what were they originally designed to do?
- Viruses seem very well suited to move information around within and between genomes and to control the expression of other genetic elements within a given genome in various ways.
- Viral sequences are currently responsible for a host of beneficial function within the human genome – some of which are vital for life or the unique level of human functionality as compared to any other species (such as the unique structure and function of the human brain and the enormous complexities of embryogenesis).
- While protein-coding genes are the basic “bricks and mortar” of the genome, non-coding sequences, such as viral-type sequences, are what control how these bricks and mortar are used and the various unique structures and functions that are formed from them. In fact, more and more evidence is coming to light that suggests that the majority of the viral-type sequences within the human genome are actually functional to one degree or another. They are what form the “blueprint”, if you will, of the structure that is being built as well as its overall function and even how it is built (i.e., embryogenesis).
- Such high level structural and functional complexity is never, ever, produced by random natural mechanisms outside of very deliberate and very intelligent design.
- While similarities are easy to explain by common descent, the functional differences within various genomes are not so easy to explain – statistically impossible, in fact, beyond the lowest levels of functional complexity.
 Could it be, then, that ERVs and other viral-type sequences were originally designed to function within the human genome (and the genomes of other species as well)? That what are now exogenous retroviruses actually originated as from endogenous retroviruses? – and not the other way around? Of course, this isn’t, exactly, an entirely new idea…
Could it be, then, that ERVs and other viral-type sequences were originally designed to function within the human genome (and the genomes of other species as well)? That what are now exogenous retroviruses actually originated as from endogenous retroviruses? – and not the other way around? Of course, this isn’t, exactly, an entirely new idea…
Human immunodeficiency virus (HIV) shares specific functionally homologous sequences with endogenous retroviruses, suggesting the possibility that recombination with ERVs could change the properties of exogenous retroviruses. Thus, ERVs may serve as a variable pool from which exogenous viruses may diversify. Exogenous retroviruses may have originated from ERVs and ERV-Ls in particular may represent an intermediate between retrotransposons and exogenous viruses. (Greenwood, et. al., 2004)
Could it be that these ERVs and other viral sequences were originally given primary control over the structure and function of genomic products? – the actual functional phenotypes that are unique to various species? If so, this would very nicely explain why many viral sequences are found in the same or similar places within species that have a similar form and function – resulting in a general nested hierarchical pattern. The inherent mobility of these viral sequences could have originally served many key functional features of high-level design – similar to modular computer programming today.
In fact, a recent paper on the Dependency Graph Hypothesis (Winston Ewert, 2018) is based on computer programming and proves a much much better fit to the genetic data found within various species as compared to the Common Descent Hypothesis (Link).
 Of course, one would also expect that the Original Design would not remain perfectly functional over time without continual maintenance – and would start to decay without the constant upkeep and maintenance of the designer. Without consistent maintenance, a gorgeous sports car will start to break down over time and will, eventually, end up as dust. The same is true of the human body. It ages and decays over time and eventually ends up as dust. And, the same is true of the human gene pool as a whole. Detrimental mutations we building up in the gene pools of all slowly reproducing species much much faster than they can be removed by natural selection (Pitman, 2018). The fact is that our entire species is inevitably devolving, headed for eventual genetic meltdown and extinction. We aren’t evolving or improving over time. The opposite is actually true. In fact, we are headed downhill so fast that there is no way we could have survived as a species long enough to have evolved from an chimp-like creature some 7 million years ago. Chimps and humans would both have gone extinct due to genetic meltdown way before now. This conclusion is also consistent with recent research strongly suggests that almost all species alive today are less than 10,000 years old – which doesn’t seem to fit at all with evolutionary expectations (Pitman, 2018).
Of course, one would also expect that the Original Design would not remain perfectly functional over time without continual maintenance – and would start to decay without the constant upkeep and maintenance of the designer. Without consistent maintenance, a gorgeous sports car will start to break down over time and will, eventually, end up as dust. The same is true of the human body. It ages and decays over time and eventually ends up as dust. And, the same is true of the human gene pool as a whole. Detrimental mutations we building up in the gene pools of all slowly reproducing species much much faster than they can be removed by natural selection (Pitman, 2018). The fact is that our entire species is inevitably devolving, headed for eventual genetic meltdown and extinction. We aren’t evolving or improving over time. The opposite is actually true. In fact, we are headed downhill so fast that there is no way we could have survived as a species long enough to have evolved from an chimp-like creature some 7 million years ago. Chimps and humans would both have gone extinct due to genetic meltdown way before now. This conclusion is also consistent with recent research strongly suggests that almost all species alive today are less than 10,000 years old – which doesn’t seem to fit at all with evolutionary expectations (Pitman, 2018).
So, yes, there are detrimental features to numerous types of viruses in the world today. However, such detrimental and parasitic features (i.e., “selfish genes”) are likely due to degenerative changes that are affecting what once was a much better functioning machine. Still, even today, if we consider things carefully enough, we can recognize some of the glory of that original design.
 0
0 0
0


 0
0
 0
0 0
0 0
0 0
0



{kind=link}