 Charles Darwin originally proposed that all living things in the world today originated from a single common ancestor from which we all share a history of common descent. This process of common descent formed the “Tree of Life”, where the process of gradual evolutionary changes over hundreds of millions of years of time produced a tree-like pattern known as a “nested hierarchical pattern”. There’s just one problem. There are a lot of inconsistencies with this pattern when it comes to real life genetic analysis of actual living things. Very different branches of the proposed tree will share very similar genetic elements that are not present in what is believed to be the most recent common ancestor(s) of the various evolutionary branches. Or certain genetic elements that are predicted to be present in a particular branch are completely missing.
Charles Darwin originally proposed that all living things in the world today originated from a single common ancestor from which we all share a history of common descent. This process of common descent formed the “Tree of Life”, where the process of gradual evolutionary changes over hundreds of millions of years of time produced a tree-like pattern known as a “nested hierarchical pattern”. There’s just one problem. There are a lot of inconsistencies with this pattern when it comes to real life genetic analysis of actual living things. Very different branches of the proposed tree will share very similar genetic elements that are not present in what is believed to be the most recent common ancestor(s) of the various evolutionary branches. Or certain genetic elements that are predicted to be present in a particular branch are completely missing.
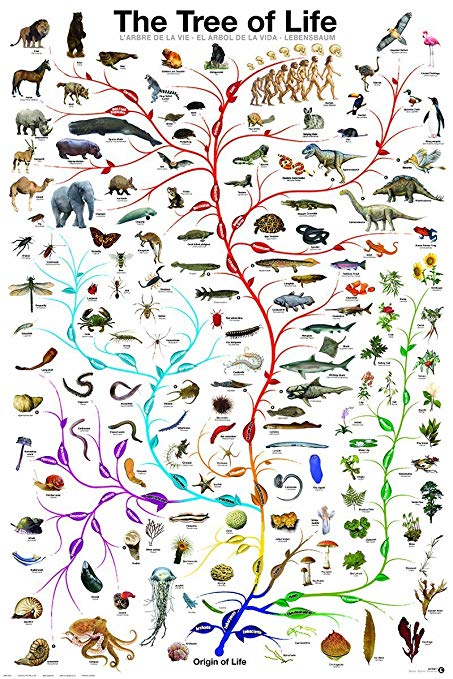
Of course, various ad hoc explanations have been forwarded in an effort to support the Tree of Life (TOL) concept, such as “horizontal gene transfer” or “convergent evolution” or “spontaneous deletion mutations”, etc. The problem is that these band-aids never really fit the evidence very well. They just don’t seem to stick properly. Still, they were and are accepted because there simply isn’t anything better to support the Darwinian story of origins.
I personally have long argued, for over 20 years now, that the information coded within the genomes of complex organisms has a striking resemblance to computer codes – which superficially resembles a nested hierarchical pattern (NHP) – with some interesting differences. Yet, there was nothing in mainstream literature to support this seemingly obvious concept – until now.
In mid-2018 Dr. Winston Ewert, a computer scientist from Baylor University with expertise in artificial intelligence, programming languages and theory of computation, published a most interesting paper on this concept entitled “The Dependency Graph of Life” in the journal Bio-Complexity (Ewert W., July 31, 2018). In this paper he proposed a new competing pattern to explain the genetic elements of life in comparison to the common descent pattern. The pattern he proposed, ironically, is based on computer software development based on “common design” rather than “common descent”. And, in comparing these well-known patterns to the common descent pattern, he discovered a much much better fit to the genetic data. In fact, the fit to the data was so dramatically superior to the common descent fit that the common descent theory is effectively falsified.
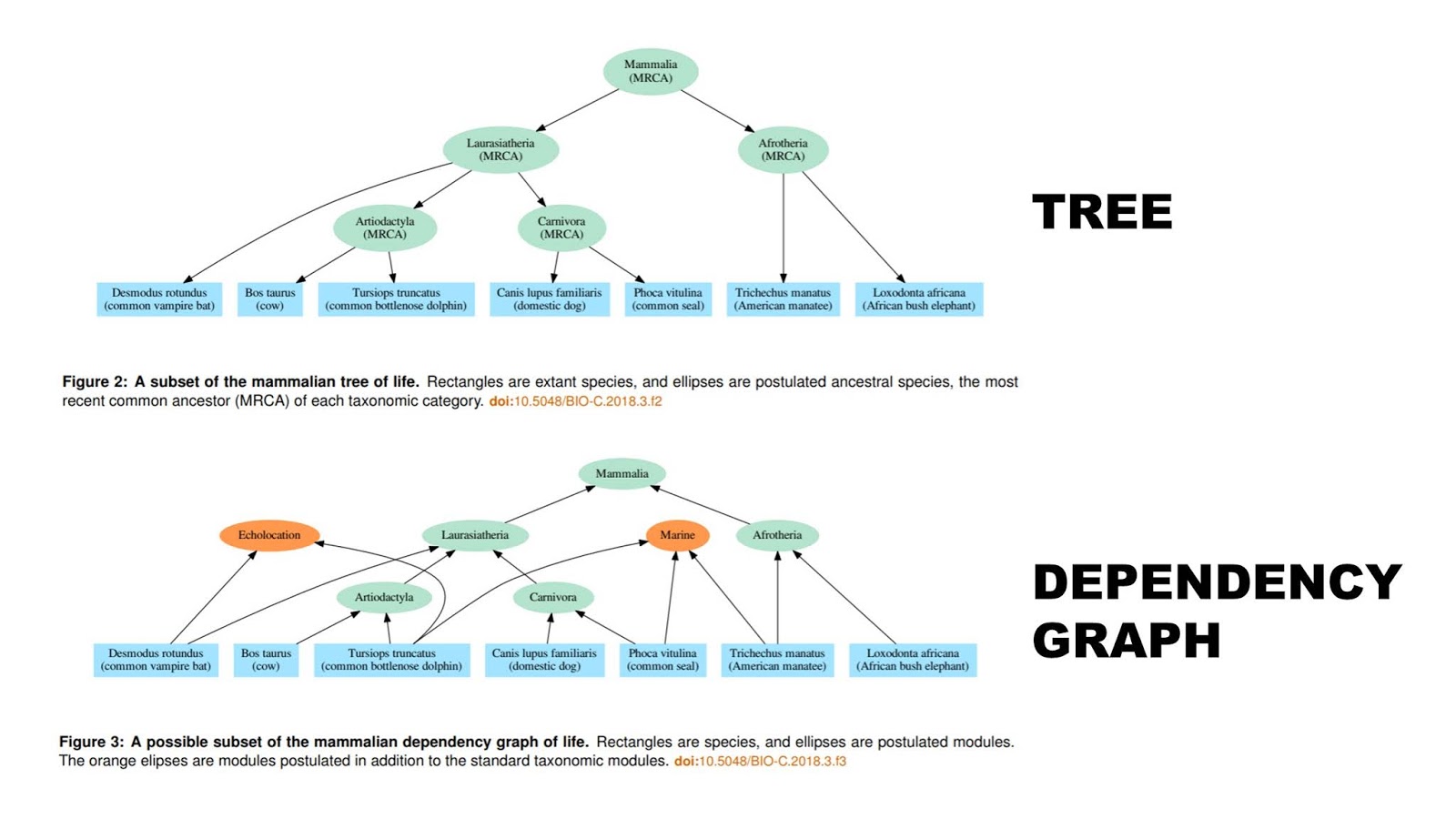
The dependency graph hypothesis derives from dependency graphs used in software development. Consequently, we can offer predictions about software produced utilizing a dependency graph. Software does not have an exact equivalent to gene families, but we can take the nearest analogue and analyze software projects in the same manner as genomes. Like the biological data, the dependency graph hypothesis predicts that software should readily fit a hierarchical pattern, and yet fit the dependency graph better than a tree…
We propose a new hypothesis to explain the approximate nested hierarchy pattern: the dependency graph. This hypothesis is drawn from the techniques used to reuse code in software development. Briefly, instead of a species descending from a single ancestral species (at least in the conventional account), each depends on multiple modules which themselves may depend on other modules. This brings together many of the ideas proposed by design-oriented critics of common descent. It uses the idea of common design by having modules reused in different species. It also draws on functional constraints, by having the valid combinations of modules restricted by which modules depend on other modules. Furthermore, it predicts an extended pattern which is the result of the proposed dependency graph mechanism…
When considering common descent, we assume a conventional understanding of common descent closer to Darwin’s “single progenitor” than to Theobald’s “population of organisms with different genotypes that lived in different places at different times.” This paper restricts its consideration to the metazoans where such a conventional understanding is still typically thought to be approximately correct. In particular, we assume that all members of a metazoan taxonomic category trace their ancestry to a particular population living at a particular time with relatively homogeneous genomes. Every new species arises by the splitting of an existing species into multiple species, leaving every species with a single ancestral species. We assume that horizontal transfer of genetic information is sufficiently rare that it can be neglected in understanding the overall pattern of metazoan life.
How much better is the fit, exactly? Well, we’re not talking about a few decimal points difference. For one of the data sets (HomoloGene), the dependency graph model was superior to common descent by a factor of 10,064. In other words, the comparison of the two models yielded a preference for the dependency graph model, as opposed to the TOL model, of greater than ten thousand. That’s astounding. The 10,064 value is the logarithm (base value of 2) of the quotient. That means that the probability of the data matching the dependency graph model is so much greater than that given the common descent model, we need logarithms even to type it out. If you tried to type out the plain number, you would have to type a 1 followed by more than 3,000 zeros!
Of course, this 10,064 bits ratio is far, far above the range where one might actually consider the competing model to be tenable. See, for example, the Bayes factor Wikipedia page (Link), showing that a Bayes factor of 3.3 bits provides “substantial” evidence for a model, 5.0 bits provides “strong” evidence, and 6.6 bits provides “decisive” evidence. If a ratio of 6.6 bits is considered to provide “decisive” evidence, consider again the decisiveness of the dependency graph model case as compared to comment descent model – with a ratio of 10,064 bits.
But this is the best case scenario for the common descent model. According to Ewert, the Bayes factor of 10,064 bits for the HomoloGene data set was the lowest ratio by a long shot. For the other eight data sets presented in the paper, the Bayes factors ranged from 40,967 to 515,450!
According to Dr. Cornelius Hunter (a graduate of the University of Illinois with a Ph.D. in Biophysics and Computational Biology, an Adjunct Professor at Biola University):
Ewert’s results are a Copernican Revolution moment. First, for the sample computer software data, not surprisingly the null model performed poorly. Computer software is highly organized, and there are relationships between different computer programs, and how they draw from foundational software libraries. But comparing the common descent and dependency graph models, the latter performs far better at modeling the software “species.” In other words, the design and development of computer software is far better described and modeled by a dependency graph than by a common descent tree…
What Ewert has shown is that, as with computer applications which inherit software from a diverse range of lower-level modules, and those lower-level modules likewise feed into a diverse range of applications, biology’s genomes likewise reveal such patterns. Genomes may inherit molecular sequence information from a wide range of genetic modules, and genetic modules may feed into a diverse range of genomes.
What about Ewert? What are his own conclusions in his paper?
If the hypothesis of common descent can explain a hierarchy, lack of a hierarchy, and the appearance of a dependency graph, what sort of predictive power can the hypothesis possibly hold? It is not enough to merely explain the features of the data, rather they must be explained within the context of a testable
hypothesis. Critiquing those rejecting common descent, White et al wrote“It is essential that any person who does not accept the continuity of evolution puts forward alternative testable models.”
This paper has answered the challenge, and paraphrasing their sentiment, it is essential that any person who does not accept the dependency graph puts forward alternative testable models. This means that it is not enough to simply postulate mechanisms to explain deviations from a hierarchy; one must postulate a hypothesis with predictions about the data…
Our key contribution is describing a proposed pattern of life expected under common design.
Common design is not restricted to finding anomalies which are difficult for common descent to explain. Many of these anomalies now fit into the overall pattern of life expected under the dependency graph. Researchers can work to understand these modules and the dependency links between them.
Our hope is that this research is a large step forward in developing common design not as a critique of common descent, but as a research program which produces testable models and increases our understanding of the biological world.
 0
0 0
0


 0
0
 0
0 0
0 0
0 0
0


