 Mark Stoeckle of Rockefeller University and David Thaler of Basel University recently published a very interesting study based on extensive analysis of over 100,000 species (“Why should mitochondria define species?”, Human Evolution, 2018). What they found shocked them. Their data showed that almost all animal species on Earth today emerged about the same time as humans.
Mark Stoeckle of Rockefeller University and David Thaler of Basel University recently published a very interesting study based on extensive analysis of over 100,000 species (“Why should mitochondria define species?”, Human Evolution, 2018). What they found shocked them. Their data showed that almost all animal species on Earth today emerged about the same time as humans.
For the planet’s 7.6 billion people, 500 million house sparrows, or 100,000 sandpipers, genetic diversity “is about the same,” Stoeckle told AFP.
“This conclusion is very surprising, and I fought against it as hard as I could,” Thaler told AFP. (Link)
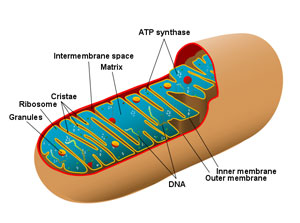 This study was based on the analysis of genetic differences within mitochondrial DNA of different species of animals. Unlike the genes in nuclear DNA, which can differ greatly from species to species, all animals have the same set of mitochondrial DNA, providing a common basis for comparison. The genetic sequences within the mitochondrial DNA can be neutral with respect to function. So, counting up the number of neutral mutational differences can be used as a clock to calculate elapsed time (given that one can determine the average mutation rate).
This study was based on the analysis of genetic differences within mitochondrial DNA of different species of animals. Unlike the genes in nuclear DNA, which can differ greatly from species to species, all animals have the same set of mitochondrial DNA, providing a common basis for comparison. The genetic sequences within the mitochondrial DNA can be neutral with respect to function. So, counting up the number of neutral mutational differences can be used as a clock to calculate elapsed time (given that one can determine the average mutation rate).
Several convergent lines of evidence show that mitochondrial diversity in modern humans follows from sequence uniformity followed by the accumulation of largely neutral diversity during a population expansion that began approximately 100,000 years ago. A straightforward hypothesis is that the extant populations of almost all animal species have arrived at a similar result consequent to a similar process of expansion from mitochondrial uniformity… (Stoeckle and Thaler, 2018).
 What is surprising here, from an evolutionary perspective anyway, is that almost all animal species have around the same number of these neutral mitochondrial mutations within their individual populations. And, the mutation rate based on phylogenic evolutionary relationships suggests that all of these tens of thousands of different species, including humans, came into existence between 100,000 and 200,000 years ago. This, by itself, is rather shocking from the Darwinian perspective. And, that’s not all. Most of these species where extremely isolated from each other, genetically, in “sequence space” with very clear genetic boundaries and nothing in between.
What is surprising here, from an evolutionary perspective anyway, is that almost all animal species have around the same number of these neutral mitochondrial mutations within their individual populations. And, the mutation rate based on phylogenic evolutionary relationships suggests that all of these tens of thousands of different species, including humans, came into existence between 100,000 and 200,000 years ago. This, by itself, is rather shocking from the Darwinian perspective. And, that’s not all. Most of these species where extremely isolated from each other, genetically, in “sequence space” with very clear genetic boundaries and nothing in between.
“If individuals are stars, then species are galaxies,” said Thaler. “They are compact clusters in the vastness of empty sequence space.”
“The absence of in-between species is something that also perplexed Darwin,” he said. (Link)


Of course, the authors specifically remarked about the “small variance within species and often but not always sequence gaps among nearest neighbor species.” In most sister species, this gap distance is in the 3-5% range, or greater, but there are many species pairs with gaps well under 1%, and variation within some species can exceed 3-5%. What this means, of course, is that the definition of a “species” is somewhat fuzzy and arbitrary. For instance, many species can interbreed to produce viable and even fertile offspring – which brings into question what it really takes to define a “species” vs. a “breed” or some kind of “ethnic variation” for instance. It is for this reason that the unique gene pools would be more accurately defined by a lack of ability to interbreed with other gene pools – which is most often the case at the genus level (with rare familial hybrids). However, as far as I’m aware, there are no known viable interordinal hybrids (though some embryonic interordinal hybrids are known).
Of course, there are a few documented interfamilial hybrids, such as between ducks (family Anatidae) and geese (family Anseridae), but maybe they shouldn’t be classified as belonging to different families. Reproductive compatibility (which is not synonymous with reproductive isolation) clearly demonstrates a close genetic relationship (such as the same number of chromosomes). It has been argued that the ability to hybridize should be limited to taxa of the same genus, which has been proposed for the classification of mammals (American Museum Novitates 2635:1-25, 1977) but not birds. So if the ability to interbreed is ever applied to taxonomy as the definition of the upper limit of the genus, there would be no such thing as an intergeneric or interfamilial hybrid. Ducks and geese would be considered to be congeneric. (Link, Link, Link)
In any case, at some point what we have are distinct “kinds” of animals with very clear genetic boundaries all coming into existence at the same time? All this is starting to sound rather designed or even Biblical… but what about the hundreds of thousands of years?
Contrary to what some might think, the mitochondrial mutation rate used here was not determined by any sort of direct analysis, but by supposed phylogenic evolutionary relationships between humans and chimps. In other words, the mutation rate was calculated based on the assumption that the theory in question was already true. This is a rather circular assumption and, as such, all results that are based on this assumption will be consistent with this assumption – like a self-fulfilling prophecy. Since the mutation rate was calculated based on previous assumptions of expected evolutionary time, then the results will automatically “confirm” those assumptions.
If one truly wishes independent confirmation of a theory, then one cannot calibrate the confirmation test by the theory, or any part of the theory, that is being tested. And yet, this is exactly what was done by scientists such as Sarich, one of the pioneers of the molecular-clock idea. Sarich began by calculating the mutation rates of various species “…whose divergence could be reliably dated from fossils.” He then applied that calibration to the chimpanzee-human split, dating that split at from five to seven million years ago. Using Sarich’s mutation calibrations, others, like Stoeckle and Thaler, apply them to their mtDNA studies, comparing “…the ratio of mitochondrial DNA divergence among humans to that between humans and chimpanzees.” By this method, scientists have calculated that the common ancestor of all modern humans, the “African Eve”, lived about 200,000 years ago. (Link)
 Obviously then, these dates, calculated from the mtDNA analysis, must match the presupposed evolutionary time scale since the calculation is based on this presupposition. The circularity of this method is inconsistent with good scientific methodology and is worthless as far as independent predictive value is concerned. The “mitochondrial clock” theory was and still is, basically, a theory within a theory in that it has no independent predictive power outside of the overarching theory of evolution. It is surprising, then, that scientists did not catch this inherent flaw earlier. Interestingly enough though, this flaw in reasoning was not detected for many years and perhaps would have remained undetected for much longer if more direct mutation-rate analyses had not been done.
Obviously then, these dates, calculated from the mtDNA analysis, must match the presupposed evolutionary time scale since the calculation is based on this presupposition. The circularity of this method is inconsistent with good scientific methodology and is worthless as far as independent predictive value is concerned. The “mitochondrial clock” theory was and still is, basically, a theory within a theory in that it has no independent predictive power outside of the overarching theory of evolution. It is surprising, then, that scientists did not catch this inherent flaw earlier. Interestingly enough though, this flaw in reasoning was not detected for many years and perhaps would have remained undetected for much longer if more direct mutation-rate analyses had not been done.
Eventually, however, scientists who study known historical families and their genetic histories (or pedigrees), started questioning the mutation rates that were based on evolutionary phylogenetic assumptions. These scientists were “stunned” to find that the mutation rate was in fact much higher than previously thought. In fact it was about 20 times higher at around one mutation every 25 to 40 generations (about 500 to 800 years for humans). It seems that in this section of the control region for mtDNA, which has about 610 base pairs, humans typically differ from one another by about 18 mutations. By simple mathematics, it follows that modern humans share a common ancestor some 300 generations back in time. If one assumes a typical generation time of about 20 years, this places the date of the common ancestor at around 6,000 years before present (Jeanson, 2015). But how could this be?! Thomas Parsons seemed just as mystified. Consider his following comments published April of 1997, in the journal Nature Genetics:
“The rate and pattern of sequence substitutions in the mitochondrial DNA (mtDNA) control region (CR) is of central importance to studies of human evolution and to forensic identity testing. Here, we report a direct measurement of the intergenerational substitution rate in the human CR. We compared DNA sequences of two CR hypervariable segments from close maternal relatives, from 134 independent mtDNA lineages spanning 327 generational events. Ten substitutions were observed, resulting in an empirical rate of 1/33 generations, or 2.5/site/Myr. This is roughly twenty-fold higher than estimates derived from phylogenetic analyses. This disparity cannot be accounted for simply by substitutions at mutational hot spots, suggesting additional factors that produce the discrepancy between very near-term and long-term apparent rates of sequence divergence. The data also indicate that extremely rapid segregation of CR sequence variants between generations is common in humans, with a very small mtDNA bottleneck. These results have implications for forensic applications and studies of human evolution.
The observed substitution rate reported here is very high compared to rates inferred from evolutionary studies. A wide range of CR substitution rates have been derived from phylogenetic studies, spanning roughly 0.025-0.26/site/Myr, including confidence intervals. A study yielding one of the faster estimates gave the substitution rate of the CR hypervariable regions as 0.118 +- 0.031/site/Myr. Assuming a generation time of 20 years, this corresponds to ~1/600 generations and an age for the mtDNA MRCA of 133,000 y.a. Thus, our observation of the substitution rate, 2.5/site/Myr, is roughly 20-fold higher than would be predicted from phylogenetic analyses. Using our empirical rate to calibrate the mtDNA molecular clock would result in an age of the mtDNA MRCA of only ~6,500 y.a., clearly incompatible with the known age of modern humans. Even acknowledging that the MRCA of mtDNA may be younger than the MRCA of modern humans, it remains implausible to explain the known geographic distribution of mtDNA sequence variation by human migration that occurred only in the last ~6,500 years.”
Parsons, Thomas J. A high observed substitution rate in the human mitochondrial DNA control region, Nature Genetics vol. 15, April 1997, pp. 363-367 (Link)
The calculation is done in the following way: Let us consider two randomly-chosen human beings. Assuming all human beings initially have identical mitochondrial DNA, after 33 generations, two such random human families will probably differ by two mutations, since there will be two separate lines of inheritance and probably one mutation along each line. After 66 generations, two randomly chosen humans will differ by about four mutations. After 100 generations, they will differ by about six mutations. After 300 generations, they will differ by about 18 mutations, which is about the observed value.
These experiments are quite concerning to evolutionists who previously calculated that the “mitochondrial eve” (who’s mitochondria is thought to be the ancestor mitochondria to all living humans) lived about 100,000 to 200,000 years ago in Africa. The new calculations, based on the above experiments, would make her a relatively young ~6,500 years old. Now, the previous notion that modern humans are up to 10,000 generations old has to be reevaluated or at least the mtDNA basis for that assumption has to be reevaluated – and it has been.
More recent direct mtDNA mutation rate studies also seem to confirm the earlier findings by Parsons and others. In an 2001 article published in the American Journal of Human Genetics, Evelyne Heyer et al., presented their findings of the mtDNA mutation rate in deep-rooted French-Canadian pedigrees.
Their findings “Confirm[ed] earlier findings of much greater mutation rates in families than those based on phylogenetic comparisons. . . For the HVI sequences, we obtained 220 generations or 6,600 years, and for the HVII sequences 275 generations or 8,250 years. Although each of these values is associated with a large variance, they both point to ~7,000-8,000 years and, therefore, to the early Neolithic as the time of expansion [mostly northern European in origin] . . . Our overall CR mutation-rate estimate of 11.6 per site per million generations . . . is higher, but not significantly different, than the value of 6.3 reported in recent the recent pedigree study of comparable size . . . In another study (Soodyall et al. 1997), no mutations were detected in 108 transmissions. On the other hand, two substitutions were observed in 81 transmissions by Howell et al. (1996), and nine substitutions were observed in 327 transmissions by Parsons et al. (1997). Combining all these data (1,729 transmissions) results in the mutation rate of 15.5 (Cl 10.3-22.1). Taking into account only those from deep-rooting pedigrees (1,321 transmissions) (Soodyall et al. 1997; Sigurdardottir et al. 2000; the present study) leads to the value of 7.9. The latter, by avoiding experimental problems with heteroplasmy, may provide a more realistic approximation of the overall mutation rate.”
Evelyn Heyer, Ewa Zietkiewicz, Andrezej Rochowski, Vania Yotova, Jack Puymirat, and Damian Labuda, Phylogenetic and Familial Estimates of Mitochondrial Substitution Rates: Study of Control Region Mutations in Deep-Rooting Pedigrees. Am. J. Hum. Genet., 69:1113-1126. 2001 (Link)
Also, consider a 2003 paper published in the Annals of Human Genetics by B. Bonne-Tamir et al. where the authors presented their results of a their study of “Maternal and Paternal Lineages” from a small isolated Samaritan community. In this paper they concluded:
“Compared with the results obtained by others on mtDNA mutation rates, our upper limit estimate of the mutation rate of 1/61 mutations per generation is in close agreement with those previously published.” [compared with the rate determined by Parsons of 1/33 generations, a rate of 1/61 is no more than double]
B. Bonne-Tamir, M. Korostishevsky, A. J. Redd, Y. Pel-Or, M. E. Kaplan and M. F. Hammer, Maternal and Paternal Lineages of the Samaritan Isolate: Mutation Rates and Time to Most Recent Common Male Ancestor, Annals of Human Genetics, Volume 67 Issue 2 Page 153 – March 2003 (Link)
One more interesting paper published in September, 2000 in the journal Science by Denver et al. is also quite interesting. These scientists reported their work with the mtDNA mutation rates of nematode worms and found that these worm’s molecular clocks actually run about “100 times faster than previously thought”.
“Extrapolating the results directly to humans is not possible, say the scientists. But their results do support recent controversial studies suggesting that the human molecular clock also runs 100 times faster than is usually thought. This may mean that estimates of divergence between chimpanzees and humans, and the emergence of modern man, happened much more recently than currently believed, says the team. ‘Our work appears to support human analyses, which have suggested a very high rate,’ says Kelley Thomas of the University of Missouri. ‘This work is relevant to humans,’ says Doug Turnbill of the institute for Human Genetics and Newcastle University, UK. ‘If the human mutation rate is faster than thought, it would have a lot of impact in looking at human disease and forensics, as well as the evolutionary rate of humans.’ . . .
Mutation rates of mtDNA in humans are usually estimated by comparing sequences of DNA from people and other animals. ‘This is kind of analysis that was used to determine that the African origin of modern humans was about 200,000 years ago,’ says Thomas. ‘The problem with this approach is that you are looking at both the mutation rate and the effects of natural selection,’ he says. The technique would also miss multiple mutations in the same stretch of mtDNA, says Paul Sharp of the Institute of Genetics at Nottingham University, UK.
More recent studies have looked at the mtDNA of people who are distantly related but share a female ancestor. This approach has revealed higher mtDNA mutation rates. But the results have not been accepted by many scientists.
Knowing the exact rate of mutation in humans is very important for forensic science and studies of genetic disease, stresses Turnbill. Forensic identification often rests on comparing samples of DNA with samples from suspected relatives. Faster human molecular clocks could complicate established exact relationships, he says.”
Denver DR, Morris K, Lynch M, Vassilieva LL, Thomas WK. High direct estimate of the mutation rate in the mitochondrial genome of Caenorhabditis elegans. Science. 2000 Sep 29;289(5488):2342-4. (Link) Also reported by: Emma Young, Running Slow, New Scientist, September 28, 2000. (Link)
Obviously then, these rates, based on more direct observations, are nowhere near those based on indirect evolutionary assumptions. This certainly does “complicate” things just a bit now doesn’t it? Isn’t it strange though that many scientists are still loath to accept these results? The bias in favor of both evolution as well as millions of years for assumed divergence times between creatures like apes and humans is so strong that changing the minds of those who hold such positions may be pretty much impossible.
For another example from a different species, direct comparisons of modern penguins with historically sequenced penguins have shown that their mtDNA mutation rates are 2 to 7 times faster than had previously been assumed through indirect methods.
Lambert, D.M., P.A. Ritchie, C.D. Millar, B. Holland, A.J. Drummond, and C. Baroni (2002) “Rates of evolution in ancient DNA from penguins.” Science 295: 2270-2273. (Link)
Certain of these problems have in fact led some scientists to stop using mitochondrial control-region sequences to reconstruct human phylogenies.
Ingman, Max, Henrik Kaessmann, Svante Paabo, and Ulf Gyllensten (2000), “Mitochondrial genome variation and the origin of modern humans,” Nature 408: 708-713. (Link)
Those scientist who continue to try and revise the molecular clock hypothesis have tried to slow down the clock by showing that some mtDNA regions mutate much more slowly than do other regions. The problem here is that such regions are obviously affected by natural selection. In other words, they are not functionally neutral with regard to selection pressures.
For example, real time experiments have shown that average mitochondrial genome mutation rates are around 6 x 10-8 mut/site/mitochondrial generation – in line with various estimates of average bacterial mutation rates (Compare with nDNA rate of 4.4 x 10-8 mut/site/human generation). With an average generation time of 45 days, that’s about 5 x 10-6 mut/site/year and 5 mut/site/myr.
This is about twice as high as Parsons’ rate of 2.5/mut/site/myr, but still about 40 to 50 times higher than rates based on phylogenetic comparisons and evolutionary assumptions. Still, this is the average rate of the entire mitochondrial genome of 16,000bp – including regions still under the restraints of natural selection. Functionally neutral regions would obviously sustain more mutations over a given span of time as compared with functionally constrained regions.
 So, the most reasonable conclusion here, it would seem, is that this data strongly favors of the claims of the Bible – claims regarding distinct non-overlapping “kinds” of animals that were recently created and which recently survived a severe bottle-neck event in the form of a catastrophic world-wide Noachian Flood that occurred just a few thousand years ago (Link). The genetic evidence that is currently in hand strongly supports that claim vs. the Darwinian story of origins where animals have existed and evolved continuously on this planet, beginning with a single common ancestor of all living things, over the course of hundreds of millions of years. That story simply doesn’t predict the genetic evidence that is in hand today. After all, the “missing links” within the genetic data are even more clear and striking than the missing links in the fossil record that “perplexed” Darwin so…
So, the most reasonable conclusion here, it would seem, is that this data strongly favors of the claims of the Bible – claims regarding distinct non-overlapping “kinds” of animals that were recently created and which recently survived a severe bottle-neck event in the form of a catastrophic world-wide Noachian Flood that occurred just a few thousand years ago (Link). The genetic evidence that is currently in hand strongly supports that claim vs. the Darwinian story of origins where animals have existed and evolved continuously on this planet, beginning with a single common ancestor of all living things, over the course of hundreds of millions of years. That story simply doesn’t predict the genetic evidence that is in hand today. After all, the “missing links” within the genetic data are even more clear and striking than the missing links in the fossil record that “perplexed” Darwin so…

 0
0 0
0


 0
0
 0
0 0
0 0
0 0
0


they will not change their minds as the have blinded themselves
Great article! Keep up the good work Sean!